RARE DISEASE DRUGS
FDA Has Steps Underway to Strengthen Coordination of Activities Supporting Drug Development
Report to Congressional Committees
November 2024
GAO-25-106774
United States Government Accountability Office
View GAO‑25‑106774. For more information, contact John E. Dicken at (202) 512-7114 or dickenj@gao.gov.
Highlights of GAO‑25‑106774, a report to congressional committees
November 2024
Rare Disease Drugs
FDA Has Steps Underway to Strengthen Coordination of Activities Supporting Drug Development
Why GAO Did This Study
Nearly one in 10 Americans have a rare disease, which is typically defined as any condition affecting fewer than 200,000 people in the United States. There are up to 10,000 rare diseases, however, only 5 percent of rare diseases have FDA-approved treatments.
The Consolidated Appropriations Act, 2023 includes a provision for GAO to examine FDA activities related to rare disease drug development and approval. This report, among other objectives, describes FDA’s strategies to help ensure reviewers have the necessary expertise and use appropriate flexibilities in their reviews, describes FDA’s programs to support rare disease drug development, and examines FDA’s efforts to coordinate its rare disease efforts.
GAO reviewed relevant statutes, regulations, and FDA documentation. GAO also interviewed FDA officials, drug sponsors of approved rare disease drugs, and patient advocacy groups representing individuals with rare diseases.
What GAO Found
The Food and Drug Administration (FDA) is responsible for determining whether drugs are safe and effective. The agency also works with drug sponsors and others to support the development of drugs to treat rare diseases, recognizing the unique challenges involved, such as assessing drug effectiveness in small patient populations. Rare disease drugs must meet the same standards as other drugs to be marketed, but FDA reviewers may apply flexibility in determining the evidence needed to meet the standards. Within FDA, staff in two of the agency’s centers—the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER)—review drug applications.
According to FDA officials, training and engagement with patient groups are among the important strategies to help ensure the agency’s staff have the expertise needed to review rare disease drug applications. Agency officials also said that a multi-layer review process helps ensure consistency among reviewers in regulatory decision-making. FDA officials said they are committed to using available flexibilities where appropriate to ensure safe and effective rare disease drugs are approved for marketing.
GAO found that FDA has 18 rare disease-specific programs—most initiated since 2019—intended to help address complexities common to rare disease drug development. Each of these programs aimed to do at least one of the following: advance understanding of the diseases, expand stakeholder engagement, or support drug development efforts.

FDA is the process of implementing a new Rare Disease Innovation Hub intended to enhance coordination between CDER and CBER and help expedite development and approval of safe and effective drugs for rare diseases. To do so, FDA will develop a cross-center strategic agenda with public input to help shape priorities and initiatives. According to agency officials, this new initiative will include the development of agencywide goals for the agency’s rare disease activities. While it is still early, this initiative holds promise for guiding the agency’s rare disease activities in a strategic and coordinated fashion. According to FDA, the new initiative will leverage each center’s rare disease-focused activities and enhance existing cross-center collaborations. Before the agency announced its new initiative, the two centers focused on developing center-specific goals for their respective rare disease activities. FDA officials said both centers plan to align their goals with agencywide goals once they are defined.
Abbreviations
|
ALS |
Amyotrophic lateral sclerosis |
|
ARC |
Accelerating Rare disease Cures Program |
|
CBER |
Center for Biologics Evaluation and Research |
|
CDER |
Center for Drug Evaluation and Research |
|
FDA |
Food and Drug Administration |
|
LEADER 3D |
Learning and Education to Advance and Empower Rare Disease Drug Developers |
|
PDUFA |
Prescription Drug User Fee Act |
|
RDEA |
Rare Disease Endpoint Advancement |
|
STAR |
Split Real Time Application Review |
|
START |
Support for clinical Trials Advancing Rare disease Therapeutics |
This is a work of the U.S. government and is not subject to copyright protection in the United States. The published product may be reproduced and distributed in its entirety without further permission from GAO. However, because this work may contain copyrighted images or other material, permission from the copyright holder may be necessary if you wish to reproduce this material separately.
November 18, 2024
The Honorable Bernard Sanders
Chair
The Honorable Bill Cassidy, M.D.
Ranking Member
Committee on Health, Education, Labor and Pensions
United States Senate
The Honorable Cathy McMorris Rodgers
Chair
The Honorable Frank Pallone, Jr.
Ranking Member
Committee on Energy and Commerce
House of Representatives
A rare disease or condition is typically defined as any disease or condition that affects fewer than 200,000 people in the United States.[1] With up to 10,000 different rare diseases having been identified, it is estimated that 30 million Americans–nearly one in 10—have one or more of them.[2] Many of these diseases are chronic, progressive (that is, worsening over time), and life threatening. They are also more difficult to diagnose and less likely to be treatable than common diseases. Rare diseases can have a substantial societal effect in terms of mortality, morbidity, and use of health care services.[3] They often disrupt work and school and can create financial hardship for patients and their families. Only about 5 percent of rare diseases have approved treatments.
The Food and Drug Administration (FDA), an agency within the Department Health and Human Services, is responsible for approving drugs and biologics for marketing in the United States.[4] To do so, FDA review teams examine evidence submitted by the drug sponsor to determine whether the product is safe and effective for its intended use.[5] Reviewers consider the quality and quantity of evidence in their decision to approve a drug for marketing, but are also permitted to apply flexibility when considering the type of data and evidence submitted. Within FDA, staff in two of the agency’s centers—the Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER)—review applications for drugs and biologics.[6] CDER regulates prescription and over-the-counter drugs, generic drugs, and some biologics, while CBER regulates most biologics, including cellular and gene therapies, vaccines, and blood and blood components for transfusion.[7]
The unique aspects of many rare diseases create scientific and other challenges to developing drugs to treat them and obtaining approval to market the drugs. For example, because the patient populations for rare diseases are relatively small, drug sponsors may face challenges recruiting sufficient numbers of participants for clinical trials to demonstrate a drug’s effectiveness. Recognizing these challenges, in addition to reviewing and approving drugs to treat rare diseases, FDA provides other support to foster the development of rare disease drugs. For example, FDA awards research grants for rare diseases, conducts outreach to patient advocacy groups about rare disease issues, implements initiatives to advance scientific research, and provides educational resources to sponsors.
The Consolidated Appropriations Act, 2023 includes a provision for us to review FDA activities related to the development and approval of rare disease drugs.[8] In this report we
1. describe the views of selected drug sponsor and patient advocacy groups regarding their experiences working with FDA on rare disease drug development;
2. describe strategies FDA uses to help ensure that staff reviewing rare disease drugs have requisite expertise and apply appropriate flexibility in their reviews;
3. describe FDA programs that are intended to support rare disease drug development; and
4. examine FDA’s efforts to coordinate its rare disease efforts.
In addition to the specific methodologies described below, we reviewed relevant laws and regulations, FDA guidance to drug sponsors, FDA’s commitment letter for the most recent reauthorization of the Prescription Drug User Fee Act (PDUFA), and other FDA documentation describing the agency’s rare disease and other drug development activities.[9] We also interviewed two national associations representing drug sponsors.
To describe the views of selected drug sponsor and patient advocacy groups regarding their experiences working with FDA on rare disease drug development, we interviewed or collected information from non-generalizable selections of (1) seven drug sponsors and (2) nine patient advocacy groups representing individuals with rare diseases. When we report statements made by both sponsor and patient advocacy groups collectively, we refer to them as “stakeholders.” The information we obtained from these entities cannot be generalized to all drug sponsors or patient advocacy groups.[10]
For the purposes of sponsor selection, we used FDA data
that identify approvals of drugs that received orphan-drug designation—a status
given by FDA to a drug that is intended to treat a rare disease.[11] We used this designation as a proxy
for rare disease drugs, as FDA officials explained that applications reviewed
and approved for drugs to treat rare diseases cannot be readily identified in
the agency’s data. We selected sponsors from among those with at least one new
drug application or biologics license application approved by FDA from January
2018 through September 2023, for novel products with orphan-drug designations.[12] Using FDA data, we selected sponsors
to provide variation across several factors, including sponsors that had (1)
experience with applications approved by CDER, CBER, or both; (2) more or less
experience seeking approval of orphan-designated drugs as determined by the
number of such drugs they had approved during our selection time frame; and
(3) experience developing drugs to treat rare diseases affecting smaller as
well as larger patient populations.
We also selected nine advocacy groups representing patients with rare diseases. These groups were composed of national organizations that focus on a wide range of diseases, as well organizations that focus on specific rare diseases or classes of rare diseases, including diseases that are more prevalent among certain racial or ethnic groups or underserved communities. One patient advocacy group represented classes of rare diseases, including those they considered “ultra rare” diseases.[13] Additional information on how we selected sponsors and patient advocacy groups is provided in appendix I.
To describe strategies FDA uses to help ensure that staff reviewing rare disease drug applications have requisite expertise and apply appropriate flexibility in their reviews, we reviewed FDA information on training. We also interviewed FDA officials, including the directors of CDER and CBER, who provide leadership and overall direction for each center. Within each center, we also interviewed FDA officials who oversee application reviews. In CDER, we interviewed the directors of four offices that oversee the review of applications and the directors of one division within each of those offices that conducts application reviews. In CBER, we interviewed the director of the Office of Therapeutic Products, which reviews the greatest number of rare disease drug applications within the center, as well as the director of its suboffice Office of Clinical Evaluation. In those interviews, we asked them about policies, procedures, and strategies FDA uses to help ensure staff have the necessary expertise to review rare disease drug applications and apply appropriate flexibility in their reviews. We also reviewed documentation when available. More information on how we selected directors is provided in appendix I.
We also obtained and analyzed information provided by FDA or available on the agency’s website on the: (1) number of applications approved during calendar years 2018 through 2023 for novel products with and without orphan-drug designation; (2) number of applications received during calendar years 2018 through 2020 for novel products with orphan-drug designation and the number of those applications that were approved by 2023, by FDA office; and (3) novel products with orphan-drug designations approved during calendar years 2020 through 2023, including those approved under the accelerated approval program.[14] To assess the reliability of this information, we interviewed FDA officials and compared the data provided by FDA to other publicly available FDA data, where available. Based on these steps, we determined that the data were sufficiently reliable for the purposes of our reporting objectives.
To describe FDA programs that are intended to support rare disease drug development, we examined FDA documentation on its rare disease-specific programs and other key FDA programs that are relevant to rare disease drug development. For example, we reviewed PDUFA performance reports on the status of the agency’s efforts towards its PDUFA commitments, Federal Register notices, and documentation describing specific program activities. We also attended or reviewed documentation associated with several FDA-sponsored public meetings, including FDA’s Rare Disease Day 2023, and FDA’s Rare Disease Day 2024. We also interviewed and obtained written responses from the agency regarding how it measures performance for its programs.
To examine FDA’s efforts to coordinate its rare disease efforts, we reviewed agency documentation, including CDER and CBER strategic plans, statutory requirements, commitment letters for PDUFA IV-VII, PDUFA performance reports, and center-specific annual reports, and interviewed FDA officials.[15] For FDA’s rare disease drug development activities, we reviewed strategic goals, objectives, and milestones, or evaluation plans, when available. We also assessed the extent to which FDA’s rare disease activities incorporate key practices for evidence-based policymaking identified in our prior work, which fall into four interrelated topic areas: (1) planning for results, (2) assessing and building evidence, (3) using evidence, and (4) fostering a culture of learning and continuous improvement.[16]
We conducted this performance audit from April 2023 to November 2024 in accordance with generally accepted government auditing standards. Those standards require that we plan and perform the audit to obtain sufficient, appropriate evidence to provide a reasonable basis for our findings and conclusions based on our audit objectives. We believe that the evidence obtained provides a reasonable basis for our findings and conclusions based on our audit objectives.
Background
Rare diseases encompass a wide range of diseases that vary in cause, age of onset, affected organs, size and composition of populations affected, and other aspects. Although rare diseases can affect people of any age, about one-half of those with a rare disease are children, according to the National Institutes of Health. Some rare diseases are also known to occur more frequently in certain racial or ethnic groups.
|
Examples of Rare Diseases Duchenne muscular dystrophy. Duchenne muscular dystrophy is disease in which a specific gene—the dystrophin gene—does not function properly, resulting in a deficiency of dystrophin—a protein involved in maintaining muscle integrity. The disease primarily affects males and results in progressive muscle wasting, with muscle weakness usually noticeable in early childhood. Most children with Duchenne muscular dystrophy use a wheelchair by their early teens. Heart and breathing problems also begin in the teen years and lead to serious, life-threatening complications. Sickle cell anemia. Sickle cell anemia is an inherited disease in which the body produces red blood cells that are sickle shaped rather than disc shaped. The abnormal cells do not last as long as normal red blood cells, causing anemia. In addition, the sickle cells can become stuck in blood vessels, blocking blood flow. This can cause strokes, infections, episodes of pain, or eye problems. In the United States, most people who have sickle cell anemia are of African ancestry or identify as Black. |
Source: GAO analysis of published literature on rare diseases. | GAO‑25‑106774
Drug Discovery, Development, and Marketing Approval
Discovering and developing new drugs is a complex, lengthy, and costly process, involving regulatory oversight by FDA at different stages. As part of its responsibilities, FDA reviews proposals for conducting human clinical trials and determines whether to approve drugs for sale in the United States. Throughout this process, sponsors may meet with FDA staff to seek advice about their specific drug development programs and to resolve issues that arise.
The process of bringing a drug to market starts with drug discovery, where researchers search for and identify new drug candidates to treat specific diseases. Next, pre-clinical testing is conducted, where drug candidates are tested in laboratories and on animals to determine whether they are likely to be safe and effective in humans. If a drug candidate is found to be promising in laboratory or animal studies, a drug sponsor may decide to test it on humans in clinical trials. Before starting clinical trials, a drug sponsor must submit an investigational new drug application to FDA that summarizes the preclinical data that has been collected, manufacturing information, and the proposed clinical trial protocols.[17]
Clinical trials are typically organized into three phases, with each successive phase involving an increasing number of human subjects. If the preclinical testing and clinical trials demonstrate the drug is safe and effective, the sponsor may choose to apply for approval to market the drug in the United States by submitting to FDA a new drug application or biologics license application. (See fig. 1.) However, few drugs make it past phase two clinical trials, as companies or the agency find early signs that the drug may not be sufficiently safe or effective, according to FDA.[18]
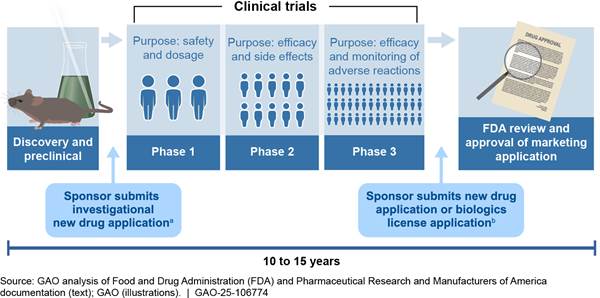
Note: This figure depicts the general steps and common time frame for drug development. Specific development steps and time frames for a particular product may vary from this example.
aSponsors must submit an investigational new drug application to FDA before human clinical trials can begin. FDA does not issue a formal approval of investigational new drug applications but can prohibit the start of a clinical trial if, for example, the agency determines that human subjects would be exposed to an unreasonable and significant risk of illness or injury.
bWhen seeking approval to market a new drug or biologic in the United States, the drug sponsor must submit a new drug application or biologics license application to FDA.
Once a sponsor submits a new drug application or biologics license application, an FDA review team—comprised of FDA reviewers such as clinicians, pharmacologists, microbiologists, and statisticians—assesses whether the studies and other evidence the sponsor submitted demonstrate that the drug is safe and effective for its intended use. To be approved, a drug’s effectiveness must be established by substantial evidence, which is defined in statute as evidence from adequate and well-controlled clinical investigations.[19] Approval also requires FDA to determine that the drug is safe for its intended use and that the benefits of the drug outweigh the risks to the patient.
If FDA determines that an application is not ready for approval, it will issue a “complete response letter” to the sponsor, describing the specific deficiencies identified and, if possible, ways to make the application viable for approval. For example, the sponsor may need to conduct additional clinical studies or address manufacturing deficiencies. The sponsor may subsequently resubmit the application for FDA’s reconsideration or choose to withdraw the application.
To help facilitate and expedite the development and review of drugs that are intended to address unmet medical needs in the treatment of serious or life-threatening diseases, FDA has several expedited programs. Although rare diseases are not the only diseases that are eligible for expedited programs, rare disease drugs may qualify for them.
These programs confer specific benefits with the potential to help reduce the development or review time needed to bring a drug to market. For example, FDA’s breakthrough therapy designation program provides sponsors with intensive guidance from FDA on efficient drug development, rolling review of application materials, and other benefits.[20] In addition, the accelerated approval program allows FDA to approve an application for a qualifying drug based on a demonstrated effect on a surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit.[21] The accelerated approval program can shorten the drug development time frame and is most often useful, according to FDA, when the disease’s course is slow, which would otherwise require a long period of time to measure the clinical benefit of the drug.[22]
Challenges of Rare Disease Drug Development and Approval
Developing rare disease drugs and meeting the standards for marketing approval is challenging due to unique aspects of rare diseases.[23] These include:
· Diseases are not well understood. According to FDA regulations, an adequate and well-controlled study is one that includes well-defined and reliable methods for assessing how an individual responds to a drug.[24] However, for many rare diseases, the signs and symptoms and the progression of the disease if left untreated—known as the “natural history” of disease—are often poorly understood, making it difficult to design and conduct clinical trials to test treatments. Without an understanding of the natural history of a disease, it is difficult to determine clinically meaningful clinical trial endpoints—events or outcomes that directly measure the effect of the drug on how a patient feels (e.g., symptom relief), functions, or survives. Additionally, some rare diseases can manifest in different ways in different people or symptoms may change over time. As a result, no single set of symptoms may be consistently associated with the disease—that is, there are multiple disease presentations. This also makes it difficult to determine the most reliable endpoints to use in a clinical trial.
· Small populations. According to FDA, random assignment of patients to treatment and control groups and double-blinding are the “gold” standard for clinical trial methodology.[25] However, the small population affected by a rare disease (some diseases affect only a few hundred or fewer individuals) can make it impractical to conduct such trials. Further, clinical trials involving a small number of participants can make it difficult to understand if the intervention was effective. That is because the larger the number of people included in the clinical trial, the better the chance to detect small but meaningful effects of the drug on the disease.
· Slow progression. FDA considers clinical outcomes—which measure whether individuals feel or function better or live longer—to be the most reliable clinical trial endpoints. However, some rare diseases are slow progressing, such that it could take years to demonstrate the effectiveness of a drug in clinical trials, which increases the time needed to develop rare disease drugs. Additionally, in the case of serious or life-threatening diseases, longer clinical trials could be detrimental to patients in a control group who would not be receiving the investigational drug during the trial.
Regulatory Flexibilities
The statutory requirements for approving drugs to treat rare diseases are the same as those for common diseases: FDA approval must be based on substantial evidence of the drug’s effectiveness and sufficient support for its safety. However, FDA regulations provide flexibility in determining the evidence needed to meet that standard.[26] Additionally, FDA guidance notes that determining whether the statutory standard of substantial evidence has been met requires an element of expert judgment.[27] FDA has acknowledged it is often more challenging for drug sponsors to develop rare disease drugs efficiently and to obtain approval for them, as well as the importance of regulatory flexibility in addressing rare disease drug development challenges. According to FDA, regulatory flexibilities include:
· Alternative clinical trial designs or number of trials. While certain clinical trial designs, such as random assignment of patients to treatment and control groups and double-blinding, are considered among the best way to assess a drug’s efficacy, FDA has stated that due to the small number of patients with rare diseases, sponsors may consider alternative clinical trial designs. This can include, for example, single-arm trials that compare outcomes from all patients who receive the same treatment to outcomes of those outside the clinical trial (e.g., from natural history study data).[28] FDA has also acknowledged that conducting a second clinical trial may not be feasible for certain rare diseases; in such cases, reliance on one adequate and well-controlled trial plus confirmatory evidence may be appropriate.[29]
· Surrogate or intermediate clinical endpoints. As previously noted, sponsors must typically show a positive effect of the drug on a clinically meaningful endpoint, such as the survival rate of patients. However, this can be challenging for rare diseases that have very slow or variable progression over years. FDA’s accelerated approval program allows FDA to approve a drug application for a qualifying drug if the drug demonstrates an effect on a surrogate or intermediate clinical endpoint reasonably likely to predict clinical benefit. For example, demonstrating that a drug can reduce protein in the urine, known as proteinuria, in patients with primary immunoglobulin A nephropathy—a rare disease that puts patients at risk of kidney failure—could be reasonably likely to predict that drug’s effectiveness in slowing loss of kidney function and reducing the risk that the patient will later experience kidney failure. For drugs approved under the accelerated approval program, sponsors are generally required to conduct additional clinical trials to confirm the predicted effect of the drugs. If the trials fail to verify the predicted effect or other clinical benefit or the applicant fails to conduct the trial with due diligence, FDA may withdraw its approval.[30]
· Uncertainty or risk. As previously noted, FDA’s approval requires the agency to determine that the benefits of the drug outweigh its risks. According to FDA guidance, the agency may be willing to accept greater uncertainty or risk—which could be due to the small number of patients included in the trial—in certain cases, such as for the treatment of serious diseases where there is unmet medical need.[31]
FDA Reviews and Approvals
Rare disease drugs comprise a substantial proportion of the novel products reviewed and approved by FDA.[32] For example, in 2023, a little over one-half of the novel drugs approved by FDA had an orphan-drug designation (see fig. 2). In total, from 2018 through 2023, 186 of the 356 novel products approved by CDER and CBER (52 percent) were for drugs that had an orphan-drug designation.
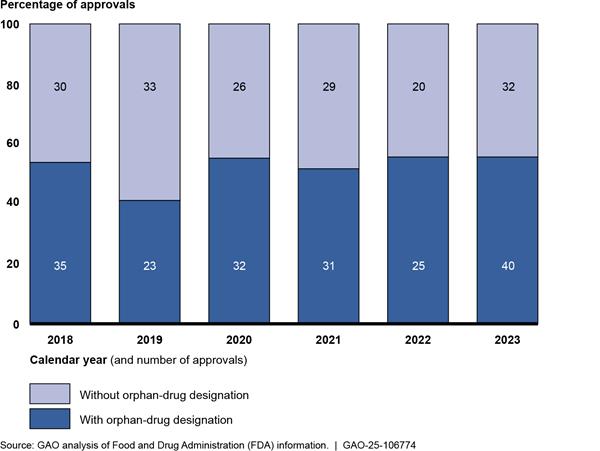
Notes: Data show the number of new drug applications and biologics license applications for novel products reviewed and approved by FDA’s Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research and their orphan-drug designation status as determined by FDA. An orphan drug is one intended to treat a rare disease, which is defined in statute as a disease that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. 21 U.S.C. 360bb(a)(2). Sponsors of rare disease drugs may submit an application to FDA for orphan-drug designation, which, if approved, entitles the sponsor to certain benefits under the Orphan Drug Act. See 21 U.S.C. 360bb(a). The orphan-drug designation process is optional for sponsors of rare disease drugs, and it is distinct from FDA’s review and approval of a new drug or biologics license application, which is required to market the drug. Novel products are therapies that have not been previously approved in the United States, including new molecular entities and novel biologics.
Applications for drugs to treat rare diseases can be reviewed by teams in any of CDER’s divisions within its Office of New Drugs, or by teams led within any of CBER’s product offices.[33] Within CBER, most reviews of rare disease drug applications are conducted by the Office of Therapeutic Products.[34]
Review teams are also supported by staff within each center that are responsible for overseeing many of the two centers’ rare disease activities, including related PDUFA commitments. Specifically, CDER’s Rare Diseases Team coordinates the development of CDER policy and training for reviewers of rare disease drug applications, among other activities. Staff from across CBER, as part of CBER’s Rare Disease Program, contribute to the development and implementation of regulatory policy and procedures relevant to biologics for rare diseases, among other activities.
Selected Stakeholders Viewed FDA’s Efforts to Engage on Rare Disease Drug Development Positively and Noted Several Challenges, Which FDA is Working to Address
FDA offers various opportunities for its stakeholders—drug sponsors and patient advocacy groups—to engage with the agency on rare disease drug development issues. Most of the seven sponsors and nine patient advocacy groups we spoke with appreciated those efforts. For example, as part of the drug development process, FDA holds patient listening sessions and patient-focused drug development meetings that seek to promote the incorporation of patient experiences and perspectives in drug development.[35]
The selected stakeholders we spoke with also identified some challenges in working with FDA. FDA has taken steps that are intended to address some of these themes, which have been raised by stakeholders to the agency through public input meetings and other forums.
Rare Disease Knowledge
Most of the sponsors we spoke with appreciated the agency’s efforts to understand rare diseases, including through the mechanisms noted above. For example, one sponsor said that meetings with FDA staff have been helpful for communicating information about the complexities of these diseases and the burden of living with them, which they said FDA staff would understandably not know because the diseases are rare. Yet most of the sponsors we spoke with noted concerns about the level of disease-specific knowledge among reviewers. Some sponsors we spoke with noted the effects this had on drug development. One sponsor gave an example of a clinical reviewer insisting that the sponsor use patient survival as an outcome measure, which the sponsor said would require a 20- to 30-year clinical trial for the slow-progressing disease.
FDA officials stated that reviewers have built a deep understanding of rare diseases gained through their review of drugs in development and have a strong commitment to finding safe and effective treatments for the rare disease community. Nonetheless, they acknowledged that given the large number of rare diseases, reviewers may not have an opportunity to become fully knowledgeable about a specific rare disease before the sponsor initiates a clinical trial. FDA officials identified strategies the agency uses to help ensure that reviewers have the necessary expertise to review rare disease drugs, which we describe later in this report.
Patient Experiences
Four patient advocacy groups and three sponsors we spoke to said that FDA has been open to engaging with patients through FDA- or externally-led engagement activities, including patient listening sessions and patient-focused drug development meetings. One patient group said that FDA actively supports patient groups in holding patient-focused drug development meetings. The patient group said these meetings are an excellent way for the patient community to educate FDA and drug developers on the patient experience and feel like FDA staff are engaged in learning.
Although most of the sponsors and patient advocacy groups we spoke with appreciated FDA’s efforts to engage with stakeholders, some stakeholders we spoke with noted related challenges. For example, another patient advocacy group said that, at the patient-focused drug development meetings, patients were unsure who at FDA was in attendance or how the information from those meetings would be disseminated to the rest of the agency. Three drug sponsors and one patient advocacy group said it is not clear how FDA considers patient experience data in the drug approval process.[36] Representatives from four patient advocacy groups said patient experience data is critical, noting instances where FDA officials did not exhibit a complete understanding of the risks patients would be willing to take, or knowledge of the existing treatment landscape.[37]
Similar comments were raised by patient group representatives and others in a December 2023 public meeting focused on rare disease patient community engagement sponsored by FDA. For example, according to a report summarizing the meeting, some participants noted the importance of reviewers being informed about and familiar with patient input received through patient listening sessions before starting the review process. Participants believed this would help reviewers better understand the conditions and preferences of the patient population.
Additionally, the report noted that a common recommendation among meeting participants was for FDA to more frequently share how the input received through patient listening sessions and patient-focused drug development meetings was used by FDA reviewers.[38] As of April 2023, patient-focused drug development program staff have shared relevant listening session summaries and related reports with the reviewers assigned to newly submitted rare disease drug applications, according to FDA officials.
Application of Flexibilities
As previously noted, FDA regulations provide flexibility in determining the evidence needed to support a determination that a drug is safe and effective. One sponsor we spoke to noted that it was encouraged by recent FDA guidance on flexibilities, and another sponsor commended statements made by the director of CBER regarding the importance of considering biomarkers, which can act as surrogates or substitutes for clinically meaningful endpoints, where relevant.
Although the agency may apply flexibilities, of the sponsors we interviewed, four cited experiences that they thought raised questions about FDA’s application of flexibilities in the drug approval process. One sponsor gave an example of one FDA center being reluctant to accept a surrogate endpoint for a disease even though the surrogate endpoint had been accepted by another center for the same disease. Additionally, two patient advocacy groups commented that FDA requiring long or randomized clinical trials with larger populations creates challenges for patients with degenerative and sometimes deadly diseases.
A February 2024 FDA report summarizing feedback from stakeholders, including sponsors, related to educational opportunities in rare disease drug development, noted that sponsors reported the process of developing and validating endpoints was not straightforward and requested FDA clarify the evidence needed to support the use of surrogate endpoints.[39] FDA has taken steps to support the development of endpoints, including by providing more opportunities for sponsors to discuss potential endpoints with the agency. However, according to FDA officials, there are circumstances where an endpoint acceptable for one product or product type might not be appropriate in other instances.
Additionally, FDA officials noted that there are unique characteristics of drugs that can result in different decisions regarding approvals and have indicated the agency is taking steps to improve communication about the different determinations. FDA officials also identified different strategies the agency uses to ensure reviewers apply appropriate flexibilities, which we describe later in this report.
Guidance
FDA has several finalized and draft guidance documents for sponsors related to rare disease drug development, including
· Rare Diseases: Early Drug Development and the Role of Pre-IND Meetings (draft issued October 2018);
· Rare Diseases: Natural History Studies for Drug Development (draft issued March 2019);
· Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products (draft issued December 2019);
· Rare Diseases: Considerations for the Development of Drugs and Biological Products (finalized December 2023);
· Real-World Evidence: Considerations Regarding Non-Interventional Studies for Drug and Biological Products (draft issued March 2024); and
· Platform Technology Designation Program for Drug Development (draft issued May 2024).
Although FDA has released several guidance documents related to rare disease drug development, five sponsors and one patient advocacy organization we spoke with said they would like FDA to develop more rare disease-specific guidance documents.[40] One of those sponsors said that it has advocated for FDA to develop guidance around biomarkers and surrogate endpoints, specifically for ultra rare diseases. Additionally, one sponsor developing a tissue-based therapy said that FDA’s guidance documents do not always provide sufficient information for working with tissue products. The sponsor said it had numerous meetings with FDA to work through the challenges of developing a drug for which there was very little precedent to follow.
One patient advocacy group said that FDA guidance documents are helpful in providing considerations and examples, however it believed that the documents can still be vague for a specific application or use case. The group pointed to the regulatory playbooks developed through FDA and the National Institutes of Health that supported the Bespoke Gene Therapy Consortium as a practical example of what information FDA guidance documents could include.[41] For example, the Consortium playbook includes a decision workflow for certain meetings at CBER, as well as a section-by-section primer for an investigational new drug application submission.
FDA officials stated that they are developing new guidance related to gene therapies and will consider other topics that could benefit from additional guidance.[42] Additionally, FDA officials said the agency created a website in 2023 to centralize guidance related to rare diseases in one place in response to stakeholder feedback.[43]
Communication
Most sponsors noted positive experiences in communicating with FDA during the drug development and application process. For example, one sponsor noted that for drugs with certain designations, such as breakthrough therapy designation, there is a lot of communication with FDA even before the application is submitted. Another sponsor described the value of FDA staff providing their preliminary comments prior to a scheduled meeting, which helps focus the discussion as the meetings typically last only 1 hour.[44]
Yet, three selected sponsors said that there is a need for regular and in-person communication with FDA throughout the drug development and application process. These sponsors said they have seen an increase in the agency’s use of written communications (known as written response only letters) in lieu of face-to-face meetings, which they said limits the quality of communication between sponsors and the agency.[45] Face-to-face discussion about biomarkers, natural history, and clinical decision-making and scientific exchange is more effective and helpful for moving rare disease drug development forward, one sponsor said. Another sponsor said that during face-to-face meetings, sponsors can hear FDA’s concerns and talk through strategies to address them.
FDA officials have acknowledged sponsors’ requests for improved communication from CDER and CBER, as well as other components within the agency, and have initiated programs to increase communication and contact with sponsors on their rare disease drug development applications. We describe these programs later in this report.
FDA Uses Various Strategies to Help Ensure Agency Reviewers Have Rare Disease Expertise and Apply Appropriate Flexibilities
Strategies to Help Ensure Expertise
FDA uses training, engagement with patient groups, and leveraging internal expertise as strategies to help ensure staff have the knowledge needed to review rare disease drug applications.
Training
FDA officials said that while each rare disease is unique, the agency focuses reviewer training on the drug development challenges that are common across rare diseases. These challenges include small patient populations and heterogeneity within diseases, which can limit clinical trial study designs. For example, to help ensure that staff have appropriate expertise on rare diseases, FDA has held an annual rare disease training day since 2011. Participation in the training day is not required, but the event is widely attended, averaging over 500 attendees the last 3 years, according to agency officials. The training is a collaborative effort between CDER and CBER and may include other components in FDA. The event provides reviewers 1-2 days of training on themes relevant to rare disease drugs, such as
· strategies for overcoming challenges in small clinical trials,
· using natural history studies to augment external controls, and
· using real-world data to inform study designs.[46]
In addition, both centers hold other rare disease trainings periodically throughout the year. For example, CBER staff are encouraged to attend monthly Review Management Update sessions that focus on various topics, such as recently issued guidance and newly implemented programs intended to help advance rare disease drug development. Additionally, since 2020, CDER has held trainings approximately quarterly, known as the Rare Disease Seminar series. These seminars are open to staff across FDA, and have averaged around 200-300 participants each, according to officials. Past seminars topics have included:
· ethical and other issues related to rare disease pediatric drug development, and
· the potential effect of new discoveries in gene editing for rare disease drug development.
FDA officials noted that CDER reviewers are required to complete a core curriculum which includes training on topics relevant to rare disease drug development, such as fundamental trial design and regulatory pathways, and flexibilities relevant to rare disease drug development, such as accelerated approval. Officials said that CBER reviewers are encouraged but not required to complete core training courses, which often include examples related to rare disease products.
Engagement with Patient Groups
FDA officials said that the agency’s reviewers also augment their expertise by learning about patients’ experiences with their diseases. An important way they said they learn about patient experiences is through patient-focused drug development meetings and other types of patient listening meetings. (See sidebar.) As previously noted, patient-focused drug development program staff now share relevant patient meeting materials with the reviewers assigned to newly submitted rare disease drug applications, according to FDA officials.[47] Reviewers also learn about patients’ diseases and experiences through established relationships with patient advocacy organizations, regular meetings with patient groups, and disease-specific conferences, officials said.
|
Patient Engagement Patient-focused drug development meetings and patient listening sessions are used by the Food and Drug Administration (FDA) to obtain patients’ perspectives on their specific diseases and needs. These meetings can be either FDA-led or patient-led. The following rare diseases were among the focus of some of these meetings in 2023 and 2024: · Atypical Hemolytic Uremic Syndrome: causes abnormal blood clots to form in small blood vessels in the kidneys, which can lead to serious medical problems if blood flow is blocked, including hemolytic anemia, thrombocytopenia, and kidney failure. · Canavan Disease: progressive, fatal, inherited disorder affecting a child’s central nervous system, muscles, and eyes; symptoms progress to include seizures, blindness, and difficulty eating and swallowing. · Guillan-Barré Syndrome: disorder in which the body's immune system attacks part of the peripheral nervous system. Symptoms include muscle weakness, numbness, and tingling sensations, which can increase in intensity to paralysis. · Juvenile Huntington’s Disease: progressive, inherited disorder affecting children or adolescents; causes breakdown of brain cells in certain areas, resulting in uncontrolled movements, loss of intellectual abilities, and emotional disturbances. Source: GAO presentation of information from FDA and the National Institutes of Health. | GAO‑25‑106774 |
Through these interactions, FDA officials said the reviewers can better understand the diversity of patients’ experiences, their symptoms, their experience with available treatments, and their ongoing unmet medical needs. These interactions can inform endpoints for clinical trials and provide important insights on patients’ willingness to accept side effects, officials said. The directors of CDER and CBER underscored the importance of reviewers engaging with patient groups for these reasons.[48]
Leveraging Internal Expertise
Consulting with, and leveraging the expertise of, other FDA staff within and among centers is another strategy FDA reviewers use to help ensure they are informed by appropriate expertise, according to officials. Staff in both centers use formal and informal consults with other internal subject matter experts. According to officials, discussions between reviews teams or across centers regarding challenging and complex issues is common, although this may not be apparent to outside stakeholders. Additionally, to ensure they have the right expertise, review teams will add staff as needed. CDER officials also said that the center’s Rare Diseases Team works to connect different divisions and offices within CDER and other centers, to consult with reviewers on rare disease-specific approaches and precedent as needed.
Further, in July 2023, CDER and CBER began an informal forum for quarterly internal reviewer discussions of rare disease products being developed. According to FDA, this new forum serves as an opportunity for CBER and CDER medical officers to interact and work more closely together on rare disease specific issues that are relevant to both centers.
By contrast, consulting with experts outside FDA was not identified as a common practice by agency officials.[49] One official in CBER said that reviewers generally do not need to consult with individual outside experts, given their ability to engage with patient groups and consult with other reviewers within the agency for a specific expertise, when needed.
According to the lead of CDER’s Rare Diseases Team, when necessary, the most common way review teams consult with outside experts during a marketing application review is through advisory committees. FDA’s 31 advisory committees—comprised of scientific experts, as well as consumer, industry, and patient representatives—provide independent expert advice to the agency on its evaluation of specific products or on general topics not related to one specific product.[50] For example, in August 2024, FDA held the first meeting of CDER’s new advisory committee on genetic metabolic diseases, which include very rare diseases that pose unique and complex drug development challenges, according to FDA. While advisory committees have the potential to bring additional expertise to FDA reviews, three stakeholders said that conflict of interest rules can pose challenges to finding rare disease experts who can serve on advisory committees.[51]
Strategies to Help Ensure Appropriate Flexibilities Used
FDA officials also cited using a variety of strategies—including internal discussion forums, multiple layers of review, and leadership expectations—to help ensure that rare disease drug application reviewers use and consistently apply appropriate flexibilities in their reviews, such as the use of accelerated approval. FDA officials also noted important factors that can affect the agency’s ability to apply flexibilities.
Internal Discussion Forums
FDA officials said that reviewers can obtain input from leadership on challenging cases, including on the use of flexibilities in reviews of rare disease drugs, through various internal forums within both centers. (See sidebar.) For example, at CDER’s Medical Policy and Program Review Council weekly meetings, divisions present issues and questions, which senior leaders discuss with other members weighing in. Rare disease drug-related issues comprised over one-third of topics discussed in fiscal years 2022 and 2023, according to officials. One director shared an example where the division discussed a rare neurodevelopmental disorder for which there were multiple drug development programs but no well-defined endpoint for the disease. Division staff sought input from the council to make sure that review teams were being consistent in the advice they were giving across drug development programs. During CBER’s monthly Rare Disease Coordinating Committee meetings, reviewers may be invited to present on specific rare disease development programs and how regulatory flexibilities are being applied, according to FDA officials.
|
Food and Drug Administration (FDA) Discussion Forums Both the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER) have their own forums for FDA reviewers to discuss drug application issues, including: CDER Medical Policy and Program Review Council: Reviewers may seek input and guidance on specific drug applications. Rare Disease Drug Development Council: Staff discuss cross-cutting rare disease issues with leaders in rare disease drug development across the Office of New Drugs and Office of Translational Sciences. CBER Medical Policy Coordinating Committee: Staff share learnings and approaches relevant to the clinical development of biologics for rare diseases, according to FDA officials. Rare Disease Coordinating Committee: Reviewers present case studies related to rare disease drug development programs and information is shared about rare disease-related policies, activities, and events. Source: GAO presentation of FDA information. | GAO‑25‑106774 |
Multiple Layers of Review
Agency officials emphasized that there is a robust review process in FDA with many layers of review and communication to ensure consistency across reviewers’ decisions, such as on clinical trial design or on marketing approval. For example, each CDER review team submits its review to the division director and deputy division director, who are responsible for ensuring quality and consistency across reviews. Additionally, within CDER, applications involving novel products are reviewed by the office director or deputy office director. One CDER office director said that supervisory reviews help ensure consistency between those divisions that see more rare disease drugs and those that do not.
Leadership Expectations
Center directors said they are committed to using available flexibilities where appropriate to bring safe and effective rare disease drugs to market and have taken steps to clarify these expectations with review teams. CDER’s director said that one of the core expectations for reviewers is to apply regulatory flexibilities to the maximum extent possible. However, CDER is a large center with many review staff, offices, and divisions, so it takes more effort to ensure teams are consistent in CDER than in CBER, the director said.[52] CDER also reviews a broader array of products than CBER. As a result, CDER has developed more formalized structures, such as the Rare Diseases Team, the Accelerating Rare disease Cures (ARC) Program, and the Medical Policy and Program Review Council, to ensure consistency and amplify division goals of applying regulatory flexibility where possible, according to officials. (See sidebar for a description of the ARC Program).
|
Accelerating Rare disease Cures (ARC) Program The ARC Program is the Center for Drug Evaluation and Research’s (CDER) center-wide collaborative effort, initiated in 2022, to provide strategic overview and coordination of CDER’s rare disease activities. Managed by CDER’s Rare Diseases Team, the ARC Program coordinates rare disease activities within the center, which include training for review staff, development of rare disease guidance documents, joint operation of pilot programs with the Center for Biologics Evaluation and Research such as the Rare Disease Endpoint Advancement Pilot Program, and working to ensure that policies and practices are shared across the center, according to Food and Drug Administration (FDA) officials. Source: GAO analysis of FDA documentation. | GAO‑25‑106774 |
CBER’s director said helping to ensure staff apply flexibilities appropriately involves center leadership disseminating knowledge through the organization, including to reviewers. Although the director noted that staff can have a natural hesitancy toward using flexibilities too much, leadership in the Office of Therapeutic Products, which reviews most of the center’s rare disease products, shares this vision of embracing the appropriate use of regulatory flexibilities for rare disease drugs and works to communicate this vision to staff.[53] Similar to CDER, CBER has a Rare Disease Coordinating Committee, where staff discuss the use of regulatory flexibilities. Additionally, officials said that CBER review staff prepare summaries of drug approvals that identify the main issues with an application, which can be helpful to develop training materials and inform conversations with sponsors to promote future consistency.
However, even as the agency employs internal discussion forums, multiple layers of review, and leadership expectations, both center directors noted that there are important factors that can affect FDA’s ability to apply regulatory flexibilities in any given situation. These factors may not always be readily apparent to the public, which could lead to perceptions that review teams are inconsistent in applying flexibilities to rare disease drug application reviews. FDA officials provided some examples of these factors:
· FDA officials emphasized that even though the challenges of rare disease drug development may be similar across many diseases, the unique features of rare diseases and drug development programs—such as the rate of disease progression, the number of patients affected, how symptoms can vary across populations, and the characteristics of the product—have to be considered when making review decisions.[54] For example, both center directors noted that the use of certain flexibilities, such as accelerated approval, may be more appropriate for products with certain characteristics. Accelerated approval, they said, may be more appropriate for gene therapies, which are reviewed by CBER, when the effect of the therapy on the targeted protein or metabolite can be measured more directly.[55] In contrast, small molecule drugs, which are reviewed by CDER, often lack the high specificity to their targets. FDA officials explained that without a well-understood mechanism for how a drug engages with the target, and evidence to support a reasonably likely benefit resulting from that engagement, the evidence to support a surrogate endpoint, and thus the use of accelerated approval, is lacking.
· CDER’s director added that the center’s ability to apply regulatory flexibilities for many rare disease drugs is dependent on making advances in understanding the biology of rare diseases. CDER’s director said that to be able to use accelerated approval or other flexibilities, such as relying on evidence from clinical trials that are smaller or more limited, translational science efforts around rare diseases must be enhanced. Translational science is the process of taking discoveries made in the laboratory, clinic, or field and transforming them into treatments or approaches that help improve health. However, the director said that CDER has seen many drug applications where drug sponsors have not done the necessary upfront work, such as developing evidence early on for biomarkers that could be leveraged for accelerated approval.
In 2023, CDER put into place a Translational Science Team, officials said, to advise review divisions on topics to engage with sponsors on as early as possible, and to support the early identification of potential biomarkers or endpoints, which are critical to understanding effects of the drug on individuals with rare diseases during clinical trials. This team is operating as a pilot initiative under the center’s ARC Program and provides consultations to review teams as requested. In May 2024, CDER officials said the Translational Science Team had worked with nine rare disease drug development programs. While this effort is intended for earlier stages of drug development, CDER officials said some of the sponsors were at later stages.
FDA Has a Variety of Programs to Advance Rare Disease Drug Development, Many of Which Were Recently Initiated
Through our review of FDA documentation and interviews with agency officials, we found that FDA has 18 rare disease-specific programs underway across CDER, CBER, and the Office of Orphan Products Development. The agency implements its programs through a variety of mechanisms, such as grants, outreach programs, pilot programs, and public-private partnerships with external entities and for which FDA’s role may vary.[56] FDA created some of these rare disease-specific drug development programs as a result of its PDUFA commitments and statutory mandates, and created others as it deemed necessary.
These programs are intended to help address several of the complexities common to rare disease drug development, such as small patient populations and lack of natural history data, according to FDA documentation. Our analysis of FDA’s rare disease-specific programs found that they have three main aims with some programs having more than one of them:
· Advancing the science underlying rare disease drug development. Our analysis found the majority of FDA’s rare disease-specific programs (15 of 18) fall into this category. The agency aims to drive the development of rare disease drugs by strengthening translational science, such as the development of endpoints or biomarkers, innovative quantitative methods, drug and disease modeling, and clinical trial design, as noted by FDA officials.[57] For example, the Critical Path for Rare Neurodegenerative Diseases is a public-private partnership that facilitates interactions and collaboration between FDA, the National Institutes of Health, and stakeholders to discuss clinical and regulatory policy issues related to a number of rare neurodegenerative diseases, such as developing and validating clinical biomarkers for Amyotrophic lateral sclerosis (ALS). Initiated in 2022 in response to a provision of the Accelerating Access to Critical Therapies for ALS Act, the program is one of nine FDA-supported public-private partnerships specific to rare diseases operated by the Critical Path Institute.[58]
· Expanding stakeholder education and outreach. We found that four of the 18 rare disease-specific programs fall into this category. Through education and outreach, FDA aims to strengthen engagement with patient advocacy groups and drug sponsors, better inform these stakeholders of the regulatory requirements for rare disease drug approval, and foster an understanding of the agency’s role and efforts in rare diseases, according to agency documentation. For example, CDER’s ARC Program staff created the Learning and Education to Advance and Empower Rare Disease Drug Developers (LEADER 3D) program in 2022 to enhance and develop educational materials to help stakeholders understand and address challenges related to bringing rare disease drugs to market. As part of the initiative, FDA sought feedback from drug sponsors and patient advocacy groups from December 2022 through April 2023 on the development and dissemination of educational materials. In February 2024, the agency issued a report that analyzed that feedback and identified several opportunities for FDA to enhance its sponsor communication and educational efforts. As of July 2024, FDA told us it was working on developing educational materials for sponsors developing rare disease drugs under this program.
· Advising rare disease drug development programs. We found that two of the 18 rare disease-specific programs fall into this category. FDA aims to support the development of novel drugs by giving sponsors the opportunity to work more closely, more often, and earlier with the agency during the drug development process, as stated by agency officials. For example, the Support for clinical Trials Advancing Rare disease Therapeutics (START) Pilot Program is a joint CDER-CBER program that intends to provide selected sponsors with more frequent advice during clinical trials through meetings and ad-hoc communication with FDA staff about drug development issues.[59] FDA first accepted applications to the program between January 2024 and March 2024, and publicly announced in June 2024 that seven sponsors were chosen from a pool of 33 requests to participate in the pilot, as told to us by agency officials. There is no set end point for this pilot. FDA will work with each selected sponsor until it reaches a pivotal development stage, such as the conclusion of its clinical trials.
In addition, the Rare Disease Endpoint Advancement (RDEA) Pilot Program is a joint CDER-CBER effort to support the development of novel endpoints, initiated in response to FDA’s commitments under the most recent PDUFA reauthorization.[60] According to FDA officials, as of June 20, 2024, CDER and CBER each selected one sponsor participant from a total of eight sponsor proposals. FDA will conduct up to four meetings with each selected sponsor to discuss appropriate endpoints for their drug development programs, with meetings occurring within 45 days of a sponsor’s request. According to FDA, on an ongoing basis, the agency plans to share lessons from the pilot with the public via presentations, guidance documents, and workshops to enhance the future use of endpoints in the rare disease space.
FDA’s rare disease programs began in 1983 with the Orphan Products Clinical Trials Grant Program. FDA has initiated most of its 18 rare disease-specific programs since 2019, including the RDEA and START pilot programs. (See fig. 3.)
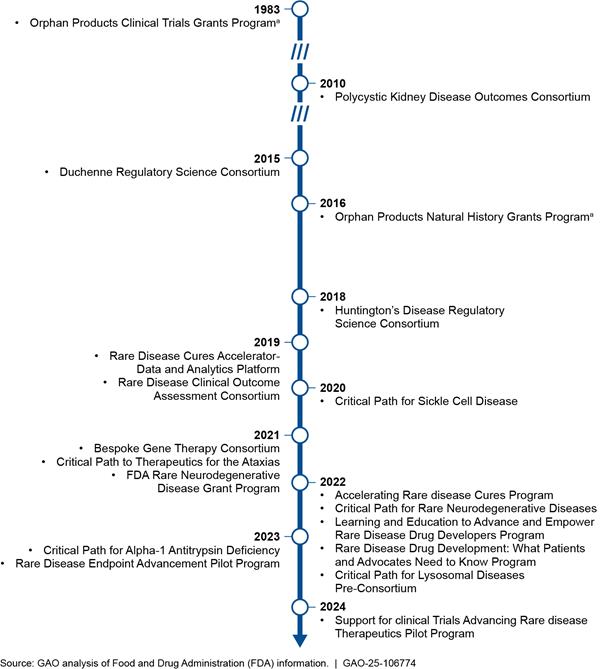
aThe Orphan Products Grant Program is comprised of the Orphan Products Clinical Trials Grants Program and the Orphan Products Natural History Grants Program, which FDA initiated in different years.
In addition to the 18 rare-disease specific programs we identified, there are several other FDA programs underway in CDER and CBER that are relevant to rare disease drug development.[61] Our analysis found that while these programs are generally aimed at supporting all drug development, they often have aims similar to FDA’s rare disease-specific programs. FDA officials identified five programs relevant for rare diseases, including the following three FDA developed in the last 2 years: the Advancing Real-World Evidence program, the Collaboration on Gene Therapies Global Pilot Program, and the Split Real Time Application Review (STAR) Pilot Program.[62] (See Appendix III for a description of the five programs.)
FDA Has Steps Underway to Enhance Coordination of Its Rare Disease Efforts
Our review shows FDA is in the process of implementing a new initiative intended to enhance coordination between CDER and CBER and help expedite development and approval of safe and effective drugs for rare diseases. Specifically, in July 2024, FDA published an announcement on the agency’s website detailing its plans to further coordinate the rare disease efforts of CDER and CBER through a Rare Disease Innovation Hub to be co-led by the directors of CDER and CBER. Through this Rare Disease Innovation Hub, FDA aims to enhance collaboration by aligning efforts across the centers to address common scientific, clinical, and policy issues for rare diseases. This enhanced collaboration is intended to promote consistency across the centers and their offices; advance regulatory science around novel rare disease drug development; and provide a single point of communication with the rare disease community.
To implement its vision for the Rare Disease Innovation Hub, FDA plans to develop a cross-center strategic agenda including to align efforts and streamline communication. As part of its initial steps in shaping the priorities and initiative for the Rare Disease Innovation Hub, FDA collected feedback from rare disease stakeholders through a public meeting held in October 2024 and written comments. In November 2024, FDA announced the agency had hired an individual to fill a new senior leadership position, known as the Director of Strategic Coalitions who will lead the development of the strategic agenda. According to CDER and CBER officials, the Rare Disease Innovation Hub will include the development of agencywide goals and appropriate progress and outcome metrics for FDA’s rare disease activities. FDA officials said no time frame had been established yet for the completion of the Innovation Hub’s strategic agenda.
FDA officials told us that before the agency announced its intent to develop a cross-center strategic agenda, CDER and CBER focused on developing center-specific goals for their respective rare disease activities and collaborating on related cross-cutting activities. In 2022, CDER defined goals for its rare disease activities as a part of its ARC Program. These goals are: (1) provide strategic direction and foster the development and use of regulatory and scientific tools to support rare disease drug development; (2) engage with external stakeholders to strengthen communication and effective collaboration; and (3) ensure CDER staff have the scientific expertise and capacity to foster a culture of innovation to advise, support, and advance successful rare disease drug development.[63] For each of these goals, CDER officials told us the center defined process measures, sub-measures, and key results that CDER staff monitor.
In January 2024, CBER began a process to develop a center-specific rare disease strategic plan, which CBER officials said will also result in strategic goals to guide the center’s rare disease efforts. CBER officials did not elaborate further on the specific goals that may result from this effort, but noted they are working to complete a draft strategic plan by the end of fiscal year 2024. CDER and CBER officials told us that once the strategic agenda developed as part of the Innovation Hub is complete, they will take steps to align their center-specific goals with agencywide goals, accordingly.
FDA’s plans to develop its Rare Disease Innovation Hub are relatively recent, and details of the plans have not yet been shared as the process has just begun, as noted by FDA officials. As the development of this new initiative continues, establishing agencywide goals and using them to guide the agency’s rare disease activities would be consistent with key practices for evidence-based policymaking that we have identified in our prior work. These practices fall into four interrelated topic areas: (1) planning for results, (2) assessing and building evidence, (3) using evidence, and (4) fostering a culture of learning and continuous improvement.[64] As illustrated in figure 4, the first three topic areas and their practices can be viewed as an iterative cycle, with the fourth area and its practices central to effectively implementing that cycle.
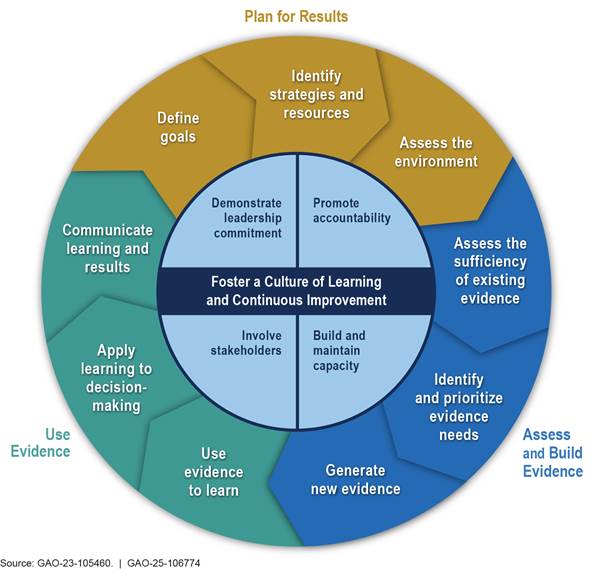
According to key practices on planning for results, establishing goals that include near-term measurable results and long-term outcomes can help an agency provide a clear picture of what it is trying to achieve and guide the organization’s activities. FDA leadership has said that setting a target for rare disease drug approvals is not an appropriate goal for the agency, given its regulatory responsibilities for ensuring that only safe and effective drugs receive marketing approval. Nonetheless, they acknowledge the importance of making measurable progress in the development of treatments to improve outcomes for patients with rare diseases.
Key practices on evidence-based policymaking also state that involving stakeholders is often vital and can help to align goals and strategies with that of others involved in achieving the same or similar outcomes. According to FDA, the agency plans to do this by obtaining public input on the priorities and initiatives for the Rare Disease Innovation Hub. Further, key practices state agencies should build evidence so they can assess progress toward goals and identify whether changes should be made to improve results. Evidence building can include developing plans to collect statistical data or to conduct an evaluation that could be used to assess the extent to which a program achieves outcomes. In the absence of agencywide goals, we found that CDER and CBER have taken steps to build evidence on the performance of many of their rare disease-specific programs. For example, under its ARC Program, CDER officials told us the center tracks the number of rare disease trainings held and number of people subscribed to the program’s listserv. They also said they plan to examine how often the advice provided through the Translational Science Team initiative is shared by review teams with sponsors and, subsequently, incorporated into sponsors’ drug applications.[65] Once FDA develops its agencywide goals, it will be important for the agency to consider the evidence it is collecting and whether that evidence will enable FDA to assess its progress toward those goals, or if additional evidence should be collected.[66]
Further, the key practices of evidence-based policymaking also show that an important part of using evidence to make informed decisions is to frequently communicate learning and results to stakeholders, tailored in a way to meet stakeholders’ needs. Doing so can help stakeholders understand how well FDA is performing and decisions the agency made to further improve results. FDA currently reports on some of its rare disease activities in multiple reports.[67] FDA officials noted they are working to develop a more comprehensive communication strategy across both centers, and that this will be a key function of the Rare Disease Innovation Hub.
While it is still early, FDA’s plan to create a new Rare Disease Innovation Hub holds promise for guiding the agency’s activities in a strategic and coordinated fashion. Continuing to incorporate key practices on evidence-based policymaking into its efforts, such as by developing a strategic agenda to include agencywide goals for its rare diseases activities, with near-term measurable results as well as long-term outcomes, could help FDA ensure this latest initiative achieves its aim of expediting the development and approval of safe and effective rare disease drugs.
Agency Comments
We provided a draft of this report to HHS for review and comment. HHS provided technical comments, which we incorporated as appropriate.
We are sending copies of this report to the appropriate congressional committees, the Secretary of Health and Human Services, the Commissioner of the Food and Drug Administration, and other interested parties. In addition, the report is available at no charge on the GAO website at http://www.gao.gov.
If you or your staff have any questions about this report, please contact me at (202) 512-7114 or dickenj@gao.gov. Contact points for our Offices of Congressional Relations and Public Affairs may be found on the last page of this report. GAO staff who made key contributions to this report are listed in appendix IV.
John E. Dicken
Director, Health Care
The following appendix provides a more detailed description of the scope and methodologies used to select and interview or collect written responses from (1) drug sponsors, (2) rare disease patient advocacy groups, and (3) directors of Food and Drug Administration (FDA) offices and divisions.[68] The information we obtained from sponsors, patient advocacy groups, and directors of FDA offices and divisions cannot be generalized to sponsors, patient advocacy groups, and FDA office directors and division directors we did not select and interview.
Drug sponsors. We interviewed and collected information from a non-generalizable sample of seven drug sponsors. To select these drug sponsors, we identified sponsors that had at least one new drug application or biologics license application approved from January 2018 through September 2023 for a novel product, that was orphan-designated.[69]
Specifically, to identify sponsors that had rare disease drugs approved by the Center for Drug Evaluation and Research (CDER), we used FDA’s Compilation of CDER New Molecular Entity Drug and New Biologic Approvals, which at the time contained approvals through calendar year 2022, and identifies orphan-designated drugs.[70] We then used FDA’s Drugs@FDA site to identify new molecular entities or novel biologics (novel products) approved during the months of January through September 2023.[71] We then determined which of those drugs were orphan-designated using an extract of data FDA provided from its Office of Orphan Product Development database.
Likewise, to identify sponsors of rare disease drugs approved by the Center for Biologics Evaluation and Research (CBER), we used the Office of Orphan Product Development database extract. We then used FDA’s website on Biological Approvals by Year to determine which drugs were approved and were novel products.[72]
Using the lists of CDER- and CBER-approved drugs with orphan-drug designations, we identified the unique sponsors—as some sponsors had more than one drug approved during the time period—and then randomized each list of sponsors. We also considered sponsors that were referred to us by advocacy organizations. Starting with sponsors at the top of the randomized lists we developed and also considering those referred to us, we selected sponsors to provide variation across several factors, including:[73]
· Center: Experience with rare disease drug application reviews by CDER, CBER, or both. Our selection of sponsors included four that had drugs approved just by CDER, one that had drugs approved just by CBER, and two that had drugs approved by both centers on at least two separate rare disease drug applications. We selected more sponsors with drugs approved by CDER because the vast majority of novel drugs with orphan designation were reviewed by CDER at the time of our review.
· Sponsor experience: We considered the level of sponsor experience with obtaining approval of rare disease drugs as determined by the number of orphan-designated drugs they had approved during the time period. Specifically, our selection of sponsors included those that had one or two orphan-designated drugs approved and those that had more than two orphan-designated drugs approved during the time period.
· Patient population: We considered sponsor experience developing drugs to treat rare diseases with smaller as well as larger populations, based on the size of the patient population a sponsor’s drug was intended to treat.[74] Specifically, we identified the estimated patient population using data provided by FDA from its Office of Orphan Product Development. Our selection of sponsors included those that had a drug approved to treat diseases affecting 5,000 or more people and those that had a drug approved to treat diseases affecting fewer than 5,000 people.
· Other factors: We also selected sponsors to ensure a mix of sponsors that had obtained marketing approvals from different review divisions within each center (which we identified by reviewing FDA application review documents) and had obtained approvals for drugs to treat diseases affecting certain populations (such as children or certain racial or ethnic groups).
Rare disease patient advocacy groups. As part of our review, we interviewed a non-generalizable selection of nine patient advocacy groups to obtain information on rare disease challenges and views regarding interactions with FDA. These groups included organizations that focus on a wide range of diseases, as well as some groups representing “ultra rare” diseases.[75] To identify the nine groups, we first selected six national groups due to their focus on patient advocacy across a broader range of diseases.
To select the other three groups, we identified rare diseases with a higher prevalence among racial or ethnic groups or underserved communities using information from prior GAO work and the EveryLife Foundation for Rare Diseases website.[76] After randomly selecting a subset of those diseases, we searched for relevant patient advocacy groups using the National Institute of Health’s Genetic and Rare Diseases Information Center and other internet searches, and reviewed FDA’s Office of Orphan Product Development database to identify diseases for which there were orphan-designated drugs—indicating drugs were being developed—but few drugs approved, which could indicate greater challenges in obtaining drug approval.[77] We also considered whether the rare disease had been the subject of a FDA patient listening or patient-focused drug development session.
FDA offices and divisions. CDER’s Office of New Drugs has eight offices with 27 review divisions that may review rare disease drug applications. We interviewed the directors of four offices and the directors of one division within each of those offices. To select these offices and divisions, we reviewed information included in FDA’s annual Prescription Drug User Fee Act (PDUFA) performance reports, which report on the status of the agency’s efforts toward commitments established under PDUFA.[78] Using the reports for fiscal years 2020 through 2022, we identified the three offices within CDER with the highest number of orphan-designated drug applications submitted. We also selected the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine, which houses the Rare Diseases Team. We then selected the division within each of the four offices that had the highest number of orphan-designated applications submitted. We conducted group interviews of directors of the following CDER offices and divisions over a 3-day period:
· Office of Cardiology, Hematology, Endocrinology and Nephrology and its Division of Non-Malignant Hematology;
· Office of Neuroscience, and its Division of Neurology I;
· Office of Oncologic Diseases, and its Division of Oncology 2; and
· Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine, and its Division of Rare Diseases and Medical Genetics.
Within CBER, we interviewed the director of the Office of Therapeutic Products, which reviews the most rare disease drug applications within CBER. Also within this office, we interviewed the director of the Office of Clinical Evaluation, who was serving as acting director of two of the three divisions within that office at the time of our review.
According to Food and Drug Administration (FDA) data on applications for orphan-drug designated novel products received by the agency from 2018 through 2020, most were approved by December 31, 2023, with minimal variation by review office within the Center for Drug Evaluation and Research (CDER) or the Center for Biologics Evaluation and Research (CBER).[79] (See fig. 5.) According to FDA officials, these data represent unique drug product applications submitted to the agency, by review office, and the number of those drug products that were subsequently approved. FDA officials also noted that the 3-year time frame after 2020 would be sufficient to capture applications that went through multiple review cycles. These data show the most recent and complete 3-year time frame at the time of our review.
This analysis has some important limitations. First, it does not capture applications submitted more recently by sponsors (i.e., those received by the agency after 2020) or indicate the number of review cycles an application may have undergone before being approved or withdrawn. This analysis also does not provide insights on sponsors’ experiences with FDA earlier in the drug development process, including experiences that may have led to delays in the sponsor submitting an application to FDA for approval or to a sponsor deciding to cease its rare disease drug development program.
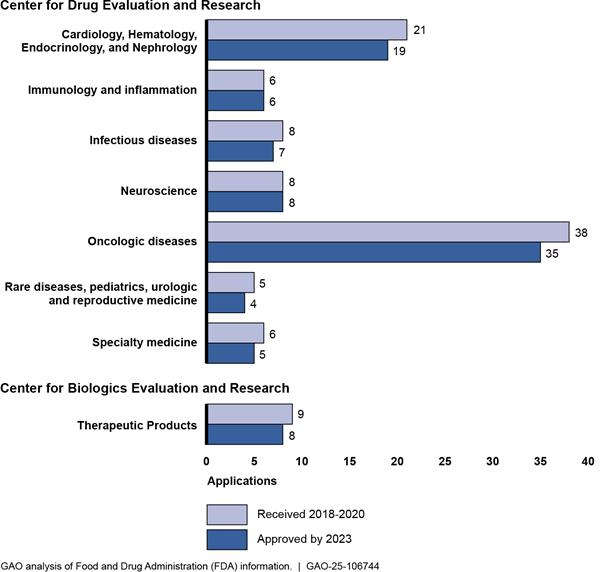
Notes: This figure shows the number of new drug applications and biologics license applications for novel products with orphan-drug designation that were received by FDA’s Center for Drug Evaluation Research (CDER) and Center for Biologics Evaluation and Research (CBER) in calendar years 2018-2020 and the number of those applications that were approved by December 31, 2023. Novel products are therapies that have not been previously approved in the United States, including new molecular entities and novel biologics. An orphan drug is one intended to treat a rare disease, which is defined in statute as a disease that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. 21 U.S.C. 360bb(a)(2). Sponsors of rare disease drugs may submit an application to FDA for orphan-drug designation, which, if approved, entitles the sponsor to certain benefits under the Orphan Drug Act. The orphan-drug designation process is optional for sponsors, and it is distinct from FDA’s review and approval of a new drug or biologics license application, which is required to market the drug. CDER and CBER completed reorganizations of their review offices in 2020 and 2023, respectively. Applications received or approved prior to the reorganizations are assigned to the current corresponding office. Offices that did not receive applications for orphan-drug designated novel products during 2018-2020 are not shown. The number of applications approved does not include applications received during 2018-2020 that were approved after 2023.
The Food and Drug Administration (FDA) has developed 18 programs specifically intended to support the development of rare disease drugs across the Center for Drug Evaluation and Research (CDER), the Center for Biologics Evaluation and Research (CBER), and the Office of Orphan Products Development. Our analysis found that the 18 programs can generally be grouped into the following three categories, although some programs have overlapping aims: expanding stakeholder education and outreach, advancing the science underlying rare disease drug development, and advising rare disease drug development programs. In addition to the 18 programs specific to rare diseases, CDER and CBER officials identified five programs that are relevant to rare diseases, but not exclusive to them. We determined these five programs have aims similar to FDA’s rare disease-specific programs.
|
Program |
Entities |
About |
Category |
|
Rare disease-specific programs |
|
||
|
Accelerating Rare disease Cures (ARC) Program |
Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER) |
Year Initiated: 2022 Description: This program is managed by CDER’s Rare Diseases Team, which facilitates and supports all of CDER’s rare disease drug development activities. The ARC Program aims to increase the development of safe and effective treatment options to address the unmet needs of patients with rare diseases by providing strategic overview and coordination of CDER’s rare disease activities. FDA officials stated that the ARC Program’s activities include developing a “one-stop shop” website for stakeholders to better access CDER’s rare disease updates, creating the Translational Science Team to support CDER staff during reviews of rare disease drug applications, and expanding outreach by hosting workshops on rare-disease specific topics. Program Measure Examples: CDER tracks the number of rare disease trainings held, the number of people subscribed to the program’s listserv, and the number of times the Translational Science Team provides advice to FDA reviewers in their review of sponsors’ drug development applications, according to agency officials. CDER plans for additional measures include conducting a qualitative assessment of whether the advice provided by the Translational Science Team was incorporated into drug development programs and led to improvements in the quality of marketing application submissions. |
Expanding stakeholder education and outreach Advancing the science underlying rare disease drug development |
|
Bespoke Gene Therapy Consortium |
FDA Center for Biologics Evaluation and Research (CBER) Foundation for the National Institutes of Health National Institutes of Health |
Year Initiated: 2021 Description: In partnership with the National Institutes of Health, FDA, and several other entities, the Bespoke Gene Therapy Consortium aims to develop streamlined models and standards that will speed the development and delivery of certain customized or “bespoke” gene therapies that could treat the millions of people affected by rare diseases. The Consortium is managed by the Foundation for the National Institutes of Health under its Accelerating Medicines Partnership program. A key deliverable was the first version of a standard operational “playbook” for developing gene therapies, which leveraged eight clinical trial test cases. Program Measure Examples: FDA officials told us the agency does not engage in any performance measurement for this program. |
Expanding stakeholder education and outreach Advancing the science underlying rare disease drug development |
|
FDA Rare Neurodegenerative Disease Grant Program |
FDA Office of Orphan Products Development |
Year Initiated: 2021 Description: FDA initiated the Rare Neurodegenerative Disease Grant Program in response to a requirement in the Accelerating Access to Critical Therapies for ALS Act.a The program awards grants and contracts to public and private entities to cover costs of research and development of interventions intended to prevent, diagnose, mitigate, treat, or cure Amyotrophic lateral sclerosis (ALS) and other rare neurodegenerative diseases in adults and children. In fiscal years 2022 and 2023, FDA awarded 11 grants and two contracts. Program Measure Examples: FDA officials told us that the Office of Orphan Products Development tracks the number of publications and collaborations created that could accelerate product development, and ways the grant helped to enhance and accelerate product development (e.g., biomarker or natural history studies funded that led to future product development). |
Advancing the science underlying rare disease drug development |
|
Learning and Education to Advance and Empower Rare Disease Drug Developers (LEADER 3D) |
FDA CDER |
Year Initiated: 2022 Description: This program is managed by CDER’s Rare Diseases Team. According to FDA officials, the purpose of the LEADER 3D initiative is to facilitate the development of safe and effective drugs to treat rare diseases through expanded stakeholder education. To facilitate this expanded stakeholder communication and education, LEADER 3D plans to develop educational materials for external stakeholders based on previously published recommendations from an analysis of stakeholder engagement, according to FDA officials. Program Measure Examples: CDER tracks the number of FDA reviewers accessing training and internal educational materials, and the number of external stakeholder materials accessed via a public website, according to agency officials. FDA officials also stated that CDER plans to develop ways to measure the effectiveness of the program, including by collecting feedback from external stakeholders on educational materials developed under the program. |
Expanding stakeholder education and outreach |
|
Orphan Products Grants Program, including: Orphan Products Clinical Trials Grants Program Orphan Products Natural History Grants Program |
FDA Office of Orphan Products Development |
Year Initiated: 1983 Description: FDA’s Orphan Products Grants Program is overseen by the Office of Orphan Products Development. The program has supported clinical research on rare diseases since 1983 and, according to agency officials, has funded clinical trials that have facilitated the approval of more than 85 products. Through the Orphan Products Clinical Trials Grants Program, FDA awards between five and 12 new clinical trial grants for orphan products annually, according to FDA officials. There are typically 60 to 85 ongoing grant projects every year. The Orphan Products Natural History Grants Program is intended to fund natural history studies that address knowledge gaps, support clinical trials and advance rare disease medical products. Through this program, FDA has awarded 16 natural history studies and has awarded new grants every two years since 2016. Program Measure Examples: According to FDA officials, the Office of Orphan Products Development tracks the number of products approved for rare diseases that received grant funding, the number of lives affected by the approvals supported by the Clinical Trials Grant Program, and publications and presentations that disseminate research results to the field. |
Advancing the science underlying rare disease drug development |
|
Rare Disease Cures Accelerator-Data and Analytics Platform |
FDA Critical Path Institute National Organization for Rare Disorders |
Year Initiated: 2019 Description: The Rare Disease Cures Accelerator-Data and Analytics Platform, funded by FDA and led by the Critical Path Institute, aims to provide a centralized and standardized infrastructure to promote the collection and sharing of rare disease patient-level data. By collecting data contributed by other organizations and making them available to other researchers and drug developers, the Rare Disease Cures Accelerator-Data and Analytics Platform aims to expedite understanding of rare disease progression and clinical outcomes measures, expediting the development of rare disease drugs. Program Measure Examples: FDA officials stated that the agency and the Critical Path Institute track data such as the number of rare diseases included on the platform, the number of data sharing agreements and individuals using data, the number of attendees at annual meetings, and the number of new drug applications or biologics license applications supported by the platform. |
Advancing the science underlying rare disease drug development |
|
Rare Disease Drug Development: What Patients and Advocates Need to Know |
FDA Critical Path Institute National Organization for Rare Disorders |
Year Initiated: 2022 Description: This program, funded by FDA and led by the National Organization for Rare Disorders, is aimed at helping patients understand the drug development process for rare diseases and encouraging patient engagement in the process. The series currently consists of an introduction and one module on the foundations of research and pre-clinical research. Future modules, to be published in 2024, include topics such as patient experience data, designing trials for small populations, and clinical trial endpoints and outcome assessments. Program Measure Examples: The National Organization for Rare Disorders tracks the number of users on the site, the amount of time spent by visitors on the site, and completion rates for the learning modules, as stated by FDA officials. |
Expanding stakeholder education and outreach |
|
Rare Diseases Endpoint Advancement (RDEA) Pilot Program |
FDA CDER FDA Center for Biologics Evaluation and Research (CBER) |
Year Initiated: 2023 Description: The RDEA Pilot Program is intended to support novel endpoint efficacy development for drugs that treat rare diseases. RDEA was initiated in response to section 3208 of the Food and Drug Omnibus Reform Act of 2022 and fulfills a commitment under the most recent reauthorization of the Prescription Drug User Fee Act (PDUFA), known as PDUFA VII.b RDEA seeks to advance rare disease drug development programs by providing a mechanism for sponsors to collaborate with FDA throughout the efficacy endpoint development process. Between fiscal years 2024 and 2027, FDA will conduct up to four meetings with each selected sponsor—with up to three sponsors selected per year—to discuss appropriate endpoints for their drug development programs. The agency also plans to share lessons from the pilot with the public via presentations, guidance documents, and workshops. Program Measure Examples: CDER and CBER track the number of proposals by endpoint type, the number of program applicants versus the number accepted, and the timelines (meeting held within 45 days of request). In addition, the centers plan to evaluate the longer-term effectiveness of the program but have not determined how they will do so, according to agency officials. For example, they may track the number of participating sponsors whose development programs go on to design and conduct a clinical trial, and the number of endpoints developed through this program that are used by another disease indication or investigational new drug application. |
Advancing the science underlying rare disease drug development Advising rare disease drug development programs |
|
Support for clinical Trials Advancing Rare disease Therapeutics (START) Pilot Program |
FDA CDER FDA CBER |
Year Initiated: 2024 Description: This joint CDER-CBER pilot will augment the currently available formal meetings between FDA and sponsors by addressing issues related to the development of individual products through more rapid, ad-hoc communication mechanisms. Sponsors selected to participate in the START Pilot Program will receive more frequent advice related to specific issues through additional interactions to facilitate novel drug development programs and generate high quality and reliable data intended to support a new drug or biologics license application. FDA will work with each selected sponsor until it reaches a pivotal development stage. Program Measure Examples: FDA officials stated that CDER and CBER will track the number of applications received versus the number accepted into the program, the number of sponsors that go on to achieve a milestone such as the initiation of a pivotal trial, and the time it took to reach a pivotal stage. In addition, FDA officials told us the centers plan to evaluate the longer-term effectiveness of the program but have not determined how they will do so. |
Advising rare disease drug development programs |
|
9 rare disease consortia led by the Critical Path Institute |
FDA Critical Path Institute |
Year Initiated: Varies Description: Supported primarily by FDA, the Critical Path Institute leads nine public-private collaborations to accelerate the development of rare disease drugs.c Eight of these collaborations are focused on specific diseases or families of diseases. These collaborations aim to increase understanding of disease progression and natural history to enhance medical advancements for each disease type by bringing together industry, academia, and regulators for discussion and knowledge development. The consortia are: 1. Critical Path for Alpha-1 Antitrypsin Deficiency, established in 2023 2. Critical Path for Lysosomal Diseases Pre-Consortium, established in 2022 3. Critical Path for Rare Neurodegenerative Diseases, established in 2022 4. Critical Path for Sickle Cell Disease, established in 2020d 5. Critical Path to Therapeutics for the Ataxias, established in 2021 6. Duchenne Regulatory Science Consortium, established in 2015 7. Huntington’s Disease Regulatory Science Consortium, established in 2018 8. Polycystic Kidney Disease Outcomes Consortium, established in 2010 In addition to these eight disease specific efforts, the Critical Path Institute leads one other collaborative effort with broader aims, the Rare Disease Clinical Outcome Assessment Consortium. This consortium was founded in 2019 and aims to accelerate the development of new drugs for rare diseases via the creation of a resource that provides information on existing clinical outcome assessments. Clinical outcome assessments are measures that describe or reflect how a patient feels, functions, or survives and could be used to support efficacy endpoints in trials for other rare disease drugs. Program Measure Examples: FDA and the Critical Path Institute track the number of membership agreements with industry representatives, patient advocates, and academic researchers founded through the consortia, as well as the number of meetings conducted by each consortium. FDA also tracks how the Critical Path Institute activities are having an effect on various aspects of drug development. |
Advancing the science underlying rare disease drug development |
|
Relevant non-rare disease-specific programs |
|
||
|
Advancing Real-World Evidence Program |
FDA CDER FDA CBER |
Year Initiated: 2022 Description: This program is a joint CDER-CBER initiative that started in 2022 to fulfill a commitment under PDUFA VII. This program provides sponsors the opportunity to meet with agency staff before protocol development or study initiation to discuss the use of real-world evidence in medical product development. Each fiscal year (2023-2027), there will be two submission cycles for applications. In 2023 and 2024, FDA will accept up to two sponsor meeting requests per submission cycle. Starting in 2025, up to four meeting requests may be accepted. Program Measure Examples: FDA officials told us that CDER and CBER track the number and type of meeting requests received and granted by each center. FDA officials also stated that CDER and CBER track the number of real-world evidence topics discussed at meetings. FDA officials told us they plan to publicly share case studies developed through the program in 2025. |
Advising rare disease drug development programs |
|
Collaboration on Gene Therapies Global Pilot Program |
FDA CBER |
Year Initiated: In early 2024, FDA announced its intention to create the Collaboration on Gene Therapies Global Pilot Program, but as of July 2024, agency officials told us they had not yet initiated the pilot program. Description: This program is intended to test the potential for simultaneous, collaborative review of gene therapy products across international regulatory partners, such as the European Medicines Agency, which will help support drug development programs globally. Program Measure Examples: As of July 2024, CBER officials indicated that this program was still in development and staff were still working to determine how to best track performance and measure effectiveness. |
Advising rare disease drug development programs |
|
Patient-Focused Drug Development Programs |
FDA CDER FDA CBER FDA Oncology Center of Excellence |
Year Initiated: 2012 Description: This program began in 2012 as part of FDA’s commitment under the fifth authorization of PDUFA.e CDER, CBER, and the Oncology Center for Excellence each have patient-focused drug development programs that work together, but also work separately on center-specific projects. The patient-focused drug development programs aim to ensure that patients’ experiences, perspectives, needs, and priorities are captured and meaningfully incorporated into drug development and evaluation. As part of this, FDA engages in FDA-led and externally-led patient-focused drug development meetings, makes condition-specific meeting reports available on a public website, and has developed guidance on the collection of patient experience data. Program Measure Examples: FDA officials stated the agency collects feedback from meeting planners following meetings and tracks the number of letters submitted by patient groups to ensure stakeholder awareness. In addition, FDA tracks the percentage of approved drug applications where review documents include patient experience data. FDA officials also noted that the agency conducted an evaluation of its externally led patient-focused drug development meetings in 2019 and 2020. |
Expanding stakeholder education and outreach Advancing the science underlying rare disease drug development |
|
Split Real Time Application Review (STAR) Pilot Program |
FDA CDER FDA CBER |
Year Initiated: 2022 Description: FDA created the STAR Pilot Program as part of its PDUFA VII commitments. The goal of this joint CDER-CBER pilot program is to shorten the time from the date of complete supplement marketing application submission to the date FDA takes its action on the submission, in order to allow earlier patient access to therapies that address an unmet medical need. Accepted applications for the program will be submitted by sponsors in two parts, approximately 2 months apart, instead of all at once, to allow for an expedited review by FDA staff. Program Measure Examples: CDER and CBER track the percentage of applications accepted into the program and the percentage of applications with an FDA regulatory action initiated within 5 months. FDA may also collect feedback on this pilot from industry stakeholders, according to agency officials. |
Advising rare disease drug development programs |
|
Standard Core Clinical Outcome Assessments and their Related Endpoints Grant Program |
FDA CDER |
Year Initiated: 2019 Description: CDER developed this grant program to support the development of a core set of clinical outcome assessments and associated endpoints for specific disease indications. This program awarded three grants in 2019 and two grants in 2021. Program Measure Examples: FDA officials told us that CDER tracks milestones, such as the development of a new clinical assessment met by grant recipients. |
Advancing the science underlying rare disease drug development |
Source: FDA information and officials. | GAO‑25‑106774
aPub. L. No. 117-79, 5, 135 Stat. 1533, 1537 (2021) (codified at 21 U.S.C. 360ee-1).
bThe Food and Drug Omnibus Reform Act of 2022 was enacted as part of the Consolidated Appropriations Act, 2023, Pub. L. No. 117-328, tit. III, 136 Stat. 4459, 5807 (2022). PDUFA VII reauthorized the prescription drug user fee program for fiscal years 2023 through 2027. See Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023, Pub. L. No. 117-180, div. F, tit. I, 136 Stat. 2114, 2139-47 (2022). For the PDUFA VII commitment letter, see Food and Drug Administration, PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2023 Through 2027, accessed June 28, 2024, https://www.fda.gov/media/151712/download.
cThe Critical Path for Rare Neurodegenerative Diseases is supported by both FDA and the National Institutes of Health.
dThe Critical Path for Sickle Cell Disease was discontinued in 2024, according to FDA officials.
ePDUFA V reauthorized the prescription drug user fee program for fiscal years 2013 through 2017. Food and Drug Administration Safety and Innovation Act, Pub. L. No. 112-144, tit. I, 126 Stat. 993, 996-1002 (2012). For the PDUFA V commitment letter, see Food and Drug Administration, PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2013 Through 2017, accessed August 8, 2024, https://www.fda.gov/media/81306/download.
GAO Contact
John E. Dicken, (202) 512-7114 or dickenj@gao.gov.
Staff Acknowledgments
In addition to the contact named above, Shannon Legeer (Assistant Director), Kendra Sippel-Theodore (Analyst-in-Charge), John Lalomio (Analyst-in-Charge), Sam Amrhein, Sonia Chakrabarty, Kaitlin Farquharson, Mairé Gebhard, Madison Herin, David Jones, Linda McIver, and Jeanne Murphy-Stone made key contributions to this report.
GAO’s Mission
The Government Accountability Office, the audit, evaluation, and investigative arm of Congress, exists to support Congress in meeting its constitutional responsibilities and to help improve the performance and accountability of the federal government for the American people. GAO examines the use of public funds; evaluates federal programs and policies; and provides analyses, recommendations, and other assistance to help Congress make informed oversight, policy, and funding decisions. GAO’s commitment to good government is reflected in its core values of accountability, integrity, and reliability.
Obtaining Copies of GAO Reports and Testimony
The fastest and easiest way to obtain copies of GAO documents at no cost is through our website. Each weekday afternoon, GAO posts on its website newly released reports, testimony, and correspondence. You can also subscribe to GAO’s email updates to receive notification of newly posted products.
Order by Phone
The price of each GAO publication reflects GAO’s actual cost of production and distribution and depends on the number of pages in the publication and whether the publication is printed in color or black and white. Pricing and ordering information is posted on GAO’s website, https://www.gao.gov/ordering.htm.
Place orders by calling (202) 512-6000, toll free (866) 801-7077,
or
TDD (202) 512-2537.
Orders may be paid for using American Express, Discover Card, MasterCard, Visa, check, or money order. Call for additional information.
Connect with GAO
Connect with GAO on Facebook, Flickr, X, and YouTube.
Subscribe to our RSS Feeds or Email Updates. Listen to our Podcasts.
Visit GAO on the web at https://www.gao.gov.
To Report Fraud, Waste, and Abuse in Federal Programs
Contact FraudNet:
Website: https://www.gao.gov/about/what-gao-does/fraudnet
Automated answering system: (800) 424-5454 or (202) 512-7700
Congressional Relations
A. Nicole Clowers, Managing Director, ClowersA@gao.gov, (202) 512-4400, U.S. Government Accountability Office, 441 G Street NW, Room 7125, Washington, DC 20548
Public Affairs
Sarah Kaczmarek, Managing Director, KaczmarekS@gao.gov, (202) 512-4800, U.S.
Government Accountability Office, 441 G Street NW, Room 7149
Washington, DC 20548
Strategic Planning and External Liaison
Stephen J. Sanford, Managing
Director, spel@gao.gov, (202) 512-4707
U.S. Government Accountability Office, 441 G Street NW, Room 7814, Washington,
DC 20548
[1]Under the Orphan Drug Act, a “rare disease or condition” is defined as one that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. Pub. L. No. 97-414, 96 Stat. 2049 (1983) (codified in relevant part, as amended, at 21 U.S.C. § 360bb(a)(2)). For the purposes of this report, we refer to diseases and conditions collectively as diseases.
[2]Estimates of the number of rare diseases are inexact, with some sources estimating a range of 7,000 to 10,000.
[3]See GAO, Rare Diseases: Although Limited, Available Evidence Suggests Medical and Other Costs Can Be Substantial, GAO‑22‑104235 (Washington, D.C.: Oct. 18, 2021), and Yang, G., et al., “The national economic burden of rare disease in the United States in 2019,” Orphanet Journal of Rare Diseases, vol. 17, no. 163 (2022).
[4]Biologics are a diverse category of products that includes vaccines and allergenic products, blood and blood components, and proteins applicable to the prevention, treatment, or cure of a disease or condition. See 42 U.S.C. § 262(i)(1). Biologics are generally derived from living material, such as the human body or a microorganism. For the purposes of this report, we refer to drugs and biologics collectively as drugs.
[5]Sponsor refers to individuals, pharmaceutical companies, or other entities that take responsibility for or initiate a clinical investigation.
[6]Though rare disease treatments can include medical devices, we did not include the Center for Devices and Radiological Health in our scope of work.
[7]CDER has responsibility for reviewing the following types of biologics: monoclonal antibodies for in vivo use; proteins intended for therapeutic use, including cytokines, enzymes, and other novel proteins, except those specifically assigned to CBER (e.g., vaccines and blood products); immunomodulators; and growth factors, cytokines, and monoclonal antibodies intended to alter the production of cells in vivo.
[8]Pub. L. No. 117-328, § 3202(f), 136 Stat. 4459, 5812-14 (2022).
[9]PDUFA authorized FDA to collect user fees from drug sponsors to support the process of reviewing drug applications. Prescription Drug User Fee Act of 1992, Pub. L. No. 102-571, tit. I, 106 Stat. 4491, 4491-4500. PDUFA has been reauthorized every 5 years since 1992; most recently, PDUFA VII reauthorized the prescription drug user fee program for fiscal years 2023 through 2027. Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023, Pub. L. No. 117-180, div. F, tit. I, 136 Stat. 2114, 2139-47 (2022).
As part of each reauthorization process, FDA identifies goals in a commitment letter to Congress. The goals in PDUFA commitment letters are the product of FDA’s discussions with the regulated industry and public stakeholders.
[10]In our report, we use the term “most” to refer to a statement made by at least half of each group—that is, at least four sponsors or at least five patient advocacy groups. We use the term “some” to refer to a statement made by two or more but less than half of each group.
[11]An orphan drug is one intended to treat a rare disease, which is defined in statute as a disease that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. 21 U.S.C. § 360bb(a)(2). Sponsors of rare disease drugs may submit an application to FDA for orphan-drug designation, which, if approved, entitles the sponsor to certain benefits under the Orphan Drug Act, such as tax credits. See 21 U.S.C. § 360bb(a). The orphan-drug designation process is optional for sponsors, and it is distinct from FDA’s review and approval of a new drug or biologics license application, which is required to market the drug. Because sponsors can choose whether to seek orphan-drug designation, orphan-designated drugs may not represent all rare disease drugs.
[12]Novel products are therapies that have not been previously approved in the United States. These include drugs classified as new molecular entities, which contain an active moiety (the core molecule or ion responsible for the physiological or pharmacological action of a drug substance) that FDA has not previously approved, and also novel biologics.
Unless otherwise noted, hereafter we use the term “application” to refer to a new drug application or a biologics license application.
[13]Though not defined in statute or regulation, the term “ultra rare” disease is used by the rare disease community to refer to a disease affecting extremely small populations. For example, some entities use the term to refer to diseases affecting fewer than 2,000 people, although definitions of ultra rare diseases vary.
Through one national patient advocacy organization, we interviewed representatives from eight patient advocacy groups that focus on ultra rare diseases. In summarizing statements made by patient advocacy organizations, we counted statements made by these representatives as from one organization.
[14]A drug may qualify for accelerated approval if it is intended to treat a serious or life-threatening disease and provides a meaningful advantage over existing treatments. 21 U.S.C. § 356(c)(1) and 21 C.F.R. § 314.500 (2024).
[15]CDER’s and CBER’s most recent strategic plans at the time of our review were Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) 2013-2017 Strategic Plan (Silver Spring, Md.: December 2, 2013); and Food and Drug Administration, Center for Biologics Evaluation and Research (CBER) 2021-2025 Strategic Plan (Silver Spring, Md.: March 19, 2021).
[16]GAO, Evidence-Based Policymaking: Practices to Help Manage and Assess the Results of Federal Efforts, GAO‑23‑105460 (Washington, D.C, July 12, 2023).
[17]FDA does not issue a formal approval to sponsors regarding investigational new drug applications. Generally, sponsors must wait 30 days to begin clinical trials after submitting their investigational new drug application. During this time, the agency can prohibit the start of a clinical trial if, for example, it determines that human subjects would be exposed to an unreasonable and significant risk of illness or injury. See 21 C.F.R. §§ 312.40 and 312.42 (2024).
[18]In addition, one study examined clinical drug development success rates in the United States from 2011 to 2020. The study found that about 52 percent of drug candidates in phase one clinical trials successfully transitioned to phase two clinical trials, and about 29 percent of those in phase two clinical trials successfully transitioned to phase three. See David Thomas et al., Clinical Development Success Rates and Contributing Factors 2011-2020, (Washington, D.C.: BIO, February 2021).
[19]Prior to a 1997 statutory amendment, FDA had interpreted the law generally to require at least two adequate and well-controlled clinical investigations to establish effectiveness. The Food and Drug Administration Modernization Act of 1997 amended the statute to provide that, if FDA determines that data from one adequate and well-controlled clinical investigation and confirmatory evidence are sufficient to establish effectiveness, FDA may consider such data and evidence to be substantial evidence when reviewing an application. Pub. L. No. 105-115, § 115(a), 111 Stat. 2296, 2313 (codified at 21 U.S.C. § 355(d)). See also 21 C.F.R. § 314.126 (2024).
[20]A drug may qualify for breakthrough therapy designation if it is intended to treat a serious or life-threatening disease and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. 21 U.S.C. § 356(a). A clinically significant endpoint is generally one that measures an effect on irreversible morbidity or mortality or on symptoms representing serious consequences of the disease. FDA can rescind breakthrough therapy designation if a drug no longer meets the qualifying criteria.
[21]A drug may qualify for accelerated approval if it is intended to treat a serious or life-threatening disease and provides a meaningful advantage over existing treatments. 21 U.S.C. § 356(c)(1) and 21 C.F.R. § 314.500 (2024). A surrogate endpoint is a marker, such as a laboratory measure or physical sign, that is reasonably likely to predict clinical benefit, but is not itself a measure of clinical benefit. For example, tumor shrinkage in certain cancer types has been considered reasonably likely to predict an improvement in, and can therefore be considered a surrogate endpoint for, overall survival. An intermediate clinical endpoint is a measurement of a therapeutic effect that can be measured earlier than an effect on irreversible morbidity or mortality and is considered reasonably likely to predict the drug’s effect on irreversible morbidity or mortality or other clinical benefit. In contrast, a clinical endpoint is a direct measure of how a patient feels, functions, or survives.
FDA generally requires that sponsors of drugs approved under the accelerated approval program conduct post-approval trials to confirm the drug’s clinical benefit. FDA may withdraw approval if the trials fail to verify the predicted clinical benefit or do not demonstrate sufficient clinical benefit to justify the risks of the drug. See 21 U.S.C. § 356(c)(3).
[22]Other examples of expedited programs include fast track designation and priority review designation. Fast track designation is intended to facilitate the development and to expedite the review of drugs intended to treat serious or life-threatening diseases that demonstrate the potential to address unmet medical needs. See 21 U.S.C. § 356(b). Priority review designation is available for drugs that are intended to treat a serious disease and, if approved, would provide a significant therapeutic improvement in safety or effectiveness.
[23]Sponsors of rare disease drugs may also face financial challenges due to the small patient populations their drugs are intended to treat. To minimize this challenge, the Orphan Drug Act provides sponsors with various incentives to develop drugs to treat rare diseases, including, for example, a tax credit for 25 percent of qualified clinical testing expenses and 7 years of exclusive marketing rights. For more information on the Orphan Drug Act and the orphan-drug designation process, see GAO, Orphan Drugs: FDA Could Improve Designation Review Consistency; Rare Disease Drug Development Challenges Continue, GAO‑19‑83 (Washington, D.C.: Nov. 30, 2018).
[24]See 21 C.F.R. § 314.126(b)(6) (2024).
[25]According to FDA, to establish a drug’s effectiveness, it is essential to distinguish the effect of the drug from other influences, including biased observations. Random assignment of trial participants to treatment and control groups and blinding are used to minimize the chance of bias by helping ensure the treatment and control groups are similar. In a double-blinded clinical trial, neither the patients nor the investigators know who is receiving the treatment. See Department of Health and Human Services, Food and Drug Administration, Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products, Guidance for Industry (Silver Spring, Md.: December 2019); and Guidance of Industry, E 10 Choice of Control Group and Related Issues in Clinical Trials (Rockville, Md.: May 2001).
[26]See 21 C.F.R. § 314.105(c) (2024).
[27]FDA guidance also notes that exercising broad scientific judgment is particularly important for life-threatening and severely debilitating diseases. See Department of Health and Human Services, Food and Drug Administration, Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products, Guidance for Industry, Draft Guidance (Silver Spring, Md.: December 2019).
[28]A natural history study is an observational study tracking the course of a disease in patients with the purpose of identifying demographic, genetic, environmental, or other variables that correlate with a disease’s development and outcomes. See Department of Health and Human Services, Food and Drug Administration, Rare Diseases: Natural History Studies for Drug Development, Guidance for Industry (Silver Spring, MD: March 2019).
[29]For relevant FDA guidance, see Department of Health and Human Services, Food and Drug Administration, Demonstrating Substantial Evidence of Effectiveness with One Adequate and Well-Controlled Clinical Investigation and Confirmatory Evidence, Guidance for Industry, Draft Guidance (Silver Spring, Md.: September 2023).
[30]The Food and Drug Omnibus Reform Act of 2022, enacted as part of the Consolidated Appropriations Act, 2023, made several changes to the accelerated approval program. Pub. L. No. 117-328, § 3210, 136 Stat. 4459, 5822 (2022). For example, it authorized FDA to require sponsors to begin confirmatory trials prior to approval, or within a specified time period after approval. 21 U.S.C. § 356(c)(2)(D). Further, if FDA does not require a drug approved under the accelerated approval program to go through a post-approval trial, FDA is required to publish on its website its rationale for why a trial is not necessary or appropriate. 21 U.S.C. § 356(c)(2)(B). In addition, the act required FDA to establish a council to ensure the consistent and appropriate use of accelerated approval across the agency. 21 U.S.C. § 356-2.
[31]See Department of Health and Human Services, Food and Drug Administration, Rare Diseases: Considerations for the Development of Drugs and Biological Products, Guidance for Industry (Silver Spring, Md.: December 2023).
[32]Novel products are therapies that have not been previously approved in the United States. These include drugs classified as new molecular entities, which contain active moieties that FDA has not previously approved, and also novel biologics.
[33]Within CDER, applications for new drugs are reviewed by staff within the Office of New Drug’s 27 review divisions, which are grouped into eight offices. Within CBER, applications are reviewed by staff, including clinical reviewers, within one of three CBER product offices. For example, within the Office of Therapeutic Products, there are six offices and 33 branches that review drug applications, with multiple offices and their divisions involved in each review.
[34]CBER has two other offices: the Office of Vaccines Research and Review and the Office of Blood Research and Review, which may also review applications for biologics intended to treat or prevent rare diseases.
[35]Patient listening sessions can either be FDA- or patient-led meetings that allow participants to inform FDA about patient experiences, perspectives, and needs related to their health or a disease. Patient listening sessions are not held to collect information about specific medical products (drug, biologic, or device) being developed. Patient-focused drug development meetings can also be FDA- or externally-led. The meetings are designed to elicit patient perspectives on their most significant symptoms, the effect of their condition on daily life, and their current approaches to treatment.
[36]Patient experience data includes data that are collected by any persons (including patients, family members and caregivers of patients, patient advocacy organizations, disease research foundations, researchers, and drug sponsors), intended to provide information about patients’ experiences with a disease. FDA reviewers may use patient experience data in their review of drug applications, including assessing the benefits and risks of the drug to patients.
Concerns about how FDA incorporates patient experience data into the drug approval process are not specific to rare disease drugs. The 21st Century Cures Act requires FDA to publish three reports on the use of patient experience data in regulatory decision-making as part of new drug and biologics license application approvals. Reports are required no later than June 1, 2021, 2026, and 2031. See Pub. L. No. 114-255, § 3004, 130 Stat. 1033, 1085 (2016). The 2021 report found that there was a lack of standardization among FDA staff in using patient experience data and that there was room for improvement. At the time, FDA said it would use the report to support its efforts to enhance the use of patient experience data in regulatory decision-making. FDA officials reported that they are taking steps to address recommendations made in the report, including as part of the agency’s commitment to advance patient-focused drug development. See Eastern Research Group, Inc. Assessment of the Use of Patient Experience Data in Regulatory Market Decision-Making (Lexington, Mass.: June 2021).
[37]In September 2024, the National Academies of Sciences, Engineering, and Medicine issued a report on the regulatory processes for rare diseases in the United States and the European Union, which made several recommendations to FDA. Among other findings, this report noted that more clarity is needed regarding how FDA uses patient input to inform regulatory decision-making and what types of patient input are the most relevant. The National Academies of Sciences, Engineering, and Medicine recommended that FDA strengthen mechanisms to incorporate input from patients with rare diseases, their caregivers, and patient representatives throughout the drug development process. See National Academies of Sciences, Engineering, and Medicine, Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities (Washington, D.C.: September 2024).
[38]The Duke-Margolis Institute for Health Policy and FDA released a report in June 2024, based on findings from a December 2023 public meeting on how to best collect and incorporate patient experience data in the rare drug development process. See Department of Health and Human Services, Food and Drug Administration and Duke-Margolis Institute for Public Health Policy, Advancing the Development of Therapeutics Through Rare Disease Patient Community Engagement (Silver Spring, Md.: June 2024).
[39]See Department of Health and Human Services, Food and Drug Administration, LEADER 3D: Learning and Education to Advance and Empower Rare Disease Drug Developers (Silver Spring, Md.: February 2024).
[40]In addition, the National Academies of Sciences, Engineering, and Medicine recommended that FDA publish or revise guidance for industry on pediatric study plans for rare disease drug development programs. See National Academies of Sciences, Engineering, and Medicine, Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
[41]The playbook provides key milestones common to a sponsor’s typical regulatory journey from research and development through clinical drug development. See Foundation for the National Institutes of Health, Bespoke Gene Therapy Consortium (BGTC) Regulatory Playbook Version 1.0 (Bethesda, Md.: February 2024).
[42]According to FDA, guidance documents, when finalized, represent the agency’s current thinking on a topic and do not create or confer any rights for or on any person and do not operate to bind FDA or the public.
[43]See https://www.fda.gov/drugs/guidances-drugs/guidance-documents-rare-disease-drug-development, accessed Aug.1, 2024.
[44]CDER and CBER have been providing written comments in advance of face-to-face meetings since 2006, according to FDA officials. They said this practice became policy in May 2009 with the publication of FDA’s draft guidance document, Formal Meetings Between FDA and Sponsors or Applicants of PDUFA Products, and was incorporated into FDA’s PDUFA VI performance goals for certain meeting types and continued with PDUFA VII. The draft guidance was updated in 2023. See Department of Health and Human Services, Food and Drug Administration, Formal Meetings Between FDA and Sponsors or Applicants of PDUFA Product (Silver Spring, Md.: September 2023).
[45]According to FDA officials in July 2024, the occurrence of written response only letters increased during the COVID-19 pandemic, but this trend is now decreasing. In a hearing before the House Committee on Energy and Commerce on May 22, 2024, the director of CBER said the center is working to reduce the number of written response only letters. Written response only letters can be requested by sponsors or be determined by FDA to be the most appropriate meeting format.
[46]A natural history study collects information about the natural history of a disease in the absence of an intervention. Its purpose is to identify variables that correlate with the disease and its outcomes, including demographic, genetic, and environmental factors. An externally controlled trial compares patients receiving the treatment to a group that is not part of the trial, as opposed to an internal control group from the same trial population. According to FDA guidance, natural history studies can be used as a comparator group in externally controlled trials. See Department of Health and Human Services, Food and Drug Administration, Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products, Guidance for Industry, Draft Guidance (Silver Spring, Md.: February 2023).
Real-world data refers to data relating to patient health status, or the delivery of health care routinely collected from a variety of sources, such as data derived from electronic health records, medical claims, and registries. See Department of Health and Human Services, Food and Drug Administration, Considerations for the Use of Real-World Data and Real-World Evidence to Support Regulatory Decision-Making for Drug and Biological Products, Guidance for Industry (Silver Spring, Md.: August 2023).
[47]Summaries of patient meetings and related materials are available on FDA’s website. For example, materials for CDER’s patient-focused drug development meetings are available at https://www.fda.gov/drugs/development-approval-process-drugs/cder-patient-focused-drug-development.
[48]FDA officials noted, however, that the agency cannot discuss information about a specific drug development program or drug application under review with patients or patient groups because that information is proprietary. FDA reviewers can only do so if sponsors bring patients or patient groups to product development or other formal FDA meetings. Some of the sponsors we interviewed said they regularly bring patient representatives to meetings with FDA while others said they generally do not do so.
[49]FDA may, when appropriate to address a specific regulatory question, consult with external experts on issues related to the review of new drugs for rare diseases, when such consultation is necessary because of a lack of specific scientific, medical, or technical expertise necessary to perform regulatory responsibilities. 21 U.S.C. § 360bbb-8(a)(2). In this context, external experts are individuals who have scientific or medical training that FDA lacks with respect to one or more rare diseases. For example, one way FDA may consult with external experts is through its Network of Experts program.
FDA is required to submit a report to congressional committees on the orphan drug program no later than September 30, 2026, including information on the extent to which the agency is consulting with external experts on rare disease drugs. Consolidated Appropriations Act, 2023, Pub. L. No. 117-328, § 3202(a), 136 Stat. at 5810. FDA did not have specific information on this at the time of our reporting, but CDER and CBER officials noted that consulting with external experts is rare.
[50]FDA may call upon an advisory committee to provide nonbinding input to the agency during the application review process.
[51]FDA advisory committee members who are special government employees are subject to federal financial conflict of interest requirements. These requirements include reporting financial information about the committee member and the member’s spouse and dependent children. FDA further reviews other financial interests related to the member, such as those of the member’s business partners or organizations for which the member serves as an officer, director, trustee, general partner, or employee.
In a hearing before the House Committee on Energy and Commerce on May 22, 2024, the director of CBER acknowledged that conflict of interest rules sometimes result in challenges to having the best possible experts participate on FDA advisory committees.
[52]In a hearing before the House Committee on Energy and Commerce on May 22, 2024, CDER’s director said that the center’s reviewers are excruciatingly aware of unmet medical needs in rare diseases and they are encouraged to exercise regulatory flexibility based on available data.
[53]CBER’s director has publicly stated interest in increasing the use of the accelerated approval program for gene therapies aimed at rare diseases. Since most rare diseases are caused by defective genes, gene therapies that correct defective genes are considered a promising treatment for rare genetic diseases.
[54]According to FDA data on applications for orphan-drug designated novel products received by the agency from 2018 through 2020, most were approved by December 31, 2023, with minimal variation by review office. See app. II for more information on this analysis and its limitations.
[55]According to FDA data, about 28 percent of novel drugs with orphan-designation approved by FDA from 2020-2023 received accelerated approval. This percentage was about 31 percent for CDER-regulated products and 13 percent for CBER-regulated products. Oncology drugs reviewed by CDER account for a large proportion of the center’s approvals using the accelerated approval program (for both orphan- and non-orphan-designated drugs). FDA officials explained that tumor shrinkage is a well-understood surrogate endpoint for many cancers, allowing for the use of accelerated approval.
[56]For example, the Orphan Products Natural History Grants Program, run by the Office of Orphan Products Development as part of the Orphan Products Grants Program, has funded 16 natural history studies to help address knowledge gaps, support clinical trials, and advance rare disease medical products since 2016.
[57]Translational science is the process of taking discoveries made in the laboratory, clinic, or field and transforming them into treatments or approaches that help improve health.
[58]Pub. L. No. 117-79, § 3, 135 Stat. 1533, 1535-36 (2021) (codified at 42 U.S.C. § 280g-7b).
The Critical Path Institute is a nonprofit aimed at facilitating scientific and regulatory pathways that accelerate the development of therapies for people with unmet medical needs. The institute operates as an independent, public-private partnership, created as an outgrowth of FDA’s 2004 Critical Path Initiative program and subsequent statutory authorization in 2007. See Food and Drug Administration Amendments Act of 2007, Pub. L. No. 110-85, § 603, 121 Stat. 823, 898 (codified, as amended, at 21 U.S.C. § 360bbb-5).
[59]Products intended to treat rare neurodegenerative diseases are eligible for potential inclusion in CDER’s START Pilot Program, and gene or cellular therapies intended to treat a rare disease that is likely to lead to significant disability or death in children are eligible for potential inclusion in CBER’s START Pilot Program.
[60]PDUFA VII reauthorized the prescription drug user fee program for fiscal years 2023 through 2027. See Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023, Pub. L. No. 117-180, div. F, tit. I, 136 Stat. 2114, 2139-47 (2022). See also Food and Drug Administration, PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2023 Through 2027, accessed June 28, 2024, https://www.fda.gov/media/151712/download. The agency will select up to three sponsors per year for this program, which will conclude in 2027.
[61]In addition to these programs, CDER and CBER also fund center-specific research development through grants. These can be rare disease-specific research but are not always. For example, CDER funded research on Seamless Design for Multicomponent Endpoints for Drug Development in Rare Diseases in 2023.
[62]In early 2024, FDA announced its intention to create the Collaboration on Gene Therapies Global Pilot Program, but as of July 2024, agency officials told us they had not yet initiated the pilot program.
[63]CDER refers to its ARC Program goals as strategic objectives. CDER has identified key efforts for its strategic objectives, and officials told us center staff meet quarterly with center leadership to review progress and consider next steps.
[64]Our prior work has identified 13 key practices that can help federal agency leaders and employees develop and use evidence to effectively manage federal performance and guide decision-making at different organizational levels, including for individual projects or programs. GAO‑23‑105460.
[65]Additionally, CDER and CBER officials told us they are currently tracking basic metrics for the RDEA and START pilot programs, such as the number of program applicants versus the number accepted. FDA officials said they plan to examine the programs’ effectiveness but have not determined how they will do so. See app. III for a description of ways FDA is measuring performance on its programs.
[66]In September 2024, the National Academies of Sciences, Engineering, and Medicine reported that most of FDA’s newer rare disease programs are limited in scope and scale compared to the scale of unmet needs in rare disease drug development. Additionally, the National Academies of Sciences, Engineering, and Medicine recommended FDA assess programs supporting rare disease drug development and publicly report those findings. See National Academies of Sciences, Engineering, and Medicine, Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
[67]FDA reports on its rare disease activities in the following reports: FDA, Accelerating Rare disease Cures (ARC) Program, Year One: Anniversary Update, June 2023; FDA, FY 2023 PDUFA Performance Report, accessed April 25, 2024, https://www.fda.gov/media/177976/download?attachment; and FDA, Center for Drug Evaluation and Research, 2023 OND Annual Report: Breaking Barrier to Advance Public Health, April 2024.
Additionally, as previously noted, FDA is required to report on the orphan drug program by September 30, 2026. Such report is to include a description of trends in rare disease drug approvals by division and the agency’s efforts to promote best practices in the development of novel rare disease drugs, among other things. See Consolidated Appropriations Act, 2023, Pub. L. No. 117-328, § 3202(a), 136 Stat. at 5810-11.
[68]Sponsor refers to individuals, pharmaceutical companies, or other entities that take responsibility for or initiate a clinical investigation.
[69]Sponsors of rare disease drugs may submit an application to FDA for orphan-drug designation, which, if approved, entitles the sponsor to certain benefits under the Orphan Drug Act, such as tax credits. See 21 U.S.C. § 360bb(a). The orphan-drug designation process is optional for sponsors, and it is distinct from FDA’s review and approval of a new drug or biologics license application, which is required to market the drug. Because sponsors of rare disease drugs can choose whether or not to seek orphan-drug designation, orphan-designated drugs may not represent all rare disease drugs.
Novel products are therapies that have not been previously approved in the United States. These include drugs classified as new molecular entities, which contain an active moiety (the core molecule or ion responsible for the physiological or pharmacological action of a drug substance) that FDA has not previously approved, and also novel biologics.
Unless otherwise noted, we use the term “application” to refer to a new drug application or a biologics license application. We also refer to both drugs and biologics as “drugs.”
[70]See https://www.fda.gov/drugs/drug-approvals-and-databases/compilation-cder-new-molecular-entity-nme-drug-and-new-biologic-approvals, accessed September 7, 2023.
[71]See https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm, accessed September 15, 2023.
[72]See https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/biological-approvals-year, accessed September 2023.
[73]As part of our selection process, we excluded sponsors that experienced mergers or acquisitions after their drugs were approved if the staff who had worked with FDA through the approval process were no longer available to speak with us. Some sponsors also declined to participate in an interview.
[74]Under the Orphan Drug Act, a “rare disease or condition” is defined as one that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. Pub. L. No. 97-414, 96 Stat. 2049 (1983) (codified in relevant part, as amended, at 21 U.S.C. § 360bb(a)(2)).
[75]Though not defined in statute or regulation, the term “ultra rare” disease is widely used to refer to a disease affecting extremely small populations. For example, some entities use the term to refer to diseases affecting fewer than 2,000 people, although definitions of ultra rare diseases vary.
Through one national patient advocacy organization, we interviewed representatives from eight patient advocacy groups that focus on ultra rare diseases. In summarizing statements made by patient advocacy organizations, we counted statements made by these representatives as from one organization.
[76]See GAO, Rare Diseases: Although Limited, Available Evidence Suggests Medical and Other Costs Can Be Substantial, GAO‑22‑104235 (Washington, D.C.: Oct. 18, 2021) and Yang, G., et al., “The national economic burden of rare disease in the United States in 2019,” Orphanet Journal of Rare Diseases, vol. 17, no. 163 (2022), https://doi.org/10.1186/s13023-022-02299-5.
[77]Sponsors may submit a request for orphan designation prior to the submission of a new drug or biologics license application. 21 U.S.C. § 360bb(a).
[78]PDUFA authorized FDA to collect user fees from drug sponsors to support the process of reviewing drug applications. Prescription Drug User Fee Act of 1992, Pub. L. No. 102-571, tit. I, 106 Stat. 4491, 4491-4500. PDUFA has been reauthorized every 5 years since 1992; most recently, PDUFA VII reauthorized the prescription drug user fee program for fiscal years 2023 through 2027. Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023, Pub. L. No. 117-180, div. F, tit. I, 136 Stat. 2114, 2139-47 (2022). As part of each reauthorization process, FDA identifies goals in a commitment letter to Congress. The goals in PDUFA commitment letters are the product of FDA’s discussions with the regulated industry and public stakeholders.
[79]Novel products are therapies that have not been previously approved in the United States. These include drugs classified as new molecular entities, which contain an active moiety (the core molecule or ion responsible for the physiological or pharmacological action of a drug substance) that FDA has not previously approved, and also novel biologics.
An orphan drug is one intended to treat a rare disease, which is defined in statute as a disease that affects fewer than 200,000 people in the United States or affects more than 200,000 and for which there is no reasonable expectation that the cost of developing and marketing the drug will be recovered from U.S. sales. 21 U.S.C. § 360bb(a)(2). Sponsors of rare disease drugs may submit an application to FDA for orphan-drug designation, which, if approved, entitles the sponsor to certain benefits under the Orphan Drug Act, such as tax credits. The orphan-drug designation process is optional for sponsors, and it is distinct from FDA’s review and approval of a new drug or biologics license application, which is required to market the drug.