DRUG SAFETY
FDA Should Implement Strategies to Retain Its Inspection Workforce
Report to Congressional Committees
November 2024
GAO-25-106775
United States Government Accountability Office
View GAO‑25‑106775. For more information, contact Mary Denigan-Macauley at (202) 512-7114 or deniganmacauleym@gao.gov.
Highlights of GAO‑25‑106775, a report to congressional committees
November 2024
DRUG SAFETY
FDA Should Implement Strategies to Retain Its Inspection Workforce
Why GAO Did This Study
FDA, an agency within the Department of Health and Human Services (HHS), inspects foreign and domestic drug manufacturers as a key oversight tool to ensure the safety and quality of drugs marketed in the U.S. In response to travel disruptions caused by the COVID-19 pandemic, FDA paused many inspections and relied on the use of alternative inspection tools to oversee drug manufacturing.
The Consolidated Appropriations Act, 2023, includes a provision for GAO to report on the status of FDA’s foreign drug inspections and its use of alternative tools. This report describes the status of FDA in-person inspections, describes FDA’s use of alternative tools, and examines FDA investigator vacancies and the agency’s efforts to address them, among other objectives. For this work, GAO examined FDA data and documents and interviewed FDA officials and investigators who conducted inspections and used alternative tools. GAO also reviewed documents from and interviewed six stakeholder groups that represent the major components of the drug manufacturing industry.
What GAO Recommends
GAO recommends that FDA collaborate to develop and implement action plans to address the remaining root causes of investigator attrition that balance inspection needs against the need to retain investigators. HHS agreed with this recommendation.
What GAO Found
After disruptions during the COVID-19 pandemic in 2020, the Food and Drug Administration (FDA) had largely resumed conducting in-person inspections of foreign and domestic drug manufacturers by March 2022. FDA data show it conducted 621 foreign and 444 domestic inspections in fiscal year 2023, although there were 36 percent fewer than in fiscal year 2019. This decrease was due in part to reduced investigator capacity, according to FDA. As of May 2024, FDA had also progressed with the implementation of two pilot programs intended to address challenges unique to foreign inspections: conducting unannounced inspections and utilizing independent interpreters.
FDA expanded the use of alternative tools to maintain oversight of drug manufacturing during the pandemic. FDA has continued to use one tool—relying on inspection reports from trusted foreign regulators—in lieu of conducting inspections. However, FDA’s use of other tools—remote assessments of information from manufacturers—has declined and will largely be reserved for more targeted use now that inspections have resumed, according to FDA.
GAO previously reported that as of November 2021, FDA had taken steps to reduce vacancies among its drug inspection workforce. However, since then, investigator attrition has generally outpaced hiring and has resulted in a large number of relatively inexperienced investigators. FDA told GAO this has limited the number of inspections FDA can complete. FDA identified root causes of attrition as the frequency and conditions of travel, pay, insufficient training, heavy workload, and issues of work-life balance. It is implementing action plans to address pay and training. FDA has not yet developed action plans to fully address travel, workload, and work-life balance because potential solutions may not allow FDA to meet its inspection needs. However, the continued loss of experienced investigators is already affecting FDA’s ability to meet inspection goals. Therefore, developing and implementing action plans to address these remaining root causes will help FDA maintain the experienced workforce it needs to oversee global drug manufacturing. This will require continued collaboration with leadership and other stakeholders to identify any actions, resources, or new authorities necessary to implement such plans.
FDA Drug Investigator Vacancies, November 2021–June
2024
|
Abbreviations |
|
|
CDER |
Center for Drug Evaluation and Research |
|
FDA |
Food and Drug Administration |
|
GMP |
Good Manufacturing Practices |
|
HHS |
Department of Health and Human Services |
|
ORA |
Office of Regulatory Affairs |
This is a work of the U.S. government and is not subject to copyright protection in the United States. The published product may be reproduced and distributed in its entirety without further permission from GAO. However, because this work may contain copyrighted images or other material, permission from the copyright holder may be necessary if you wish to reproduce this material separately.
November 13, 2024
The Honorable Bernard Sanders
Chair
The Honorable Bill Cassidy, M.D.
Ranking Member
Committee on Health, Education, Labor and Pensions
United States Senate
The Honorable Cathy McMorris Rodgers
Chair
The Honorable Frank Pallone, Jr.
Ranking Member
Committee on Energy and Commerce
House of Representatives
The Food and Drug Administration (FDA), an agency within the Department of Health and Human Services (HHS), is responsible for ensuring that drugs marketed in the U.S. are safe and effective.[1] Critical to this oversight are its inspections of the establishments manufacturing those drugs. These inspections can identify manufacturing deficiencies, which can lead to serious problems if they are not corrected. For example, in December 2022, FDA began investigating an outbreak of antibiotic-resistant bacterial infections from contaminated eye drops that led to cases of vision loss and death. An FDA inspection of the establishment identified multiple manufacturing deficiencies, including a failure to follow procedures to prevent microbiological contamination.
FDA’s inspection responsibilities have been complicated by a manufacturing supply chain that has become increasingly global. As of September 2023, 58 percent of establishments registered with FDA to manufacture drugs for the U.S. market were located overseas.[2] Over many years, FDA has increased the number of inspections it conducted and significantly increased the portion conducted overseas. By 2015, FDA was conducting more inspections in foreign countries than in the U.S.
However, as we previously reported, the COVID-19 pandemic significantly disrupted FDA’s ability to inspect drug manufacturing establishments in person.[3] Beginning in March 2020, FDA largely paused foreign and domestic drug inspections.[4] In response to these disruptions, FDA relied on the use of alternative inspection tools. Such tools included the review of inspection reports from other trusted foreign regulators and the remote assessment of information from manufacturing establishments. While FDA generally had the authority to use these tools prior to the pandemic, it had not used them extensively.
Even before the COVID-19 pandemic disruption, FDA faced challenges in conducting these inspections, which we have reported on extensively and which contribute to the inclusion of FDA’s oversight of medical products in our High-Risk series.[5] One persistent challenge has been vacancies among the investigators who conduct these inspections. Over multiple reports, we have reported on vacancies among FDA’s investigators. In December 2019, we reported that these vacancies contributed to decreased foreign and domestic inspections.[6] In 2022, we reported that FDA had undertaken a number of initiatives to recruit new investigators and had reduced these vacancies, though vacancies still existed among those investigators who specialize in foreign inspections.[7]
The Consolidated Appropriations Act, 2023, includes a provision for us to report on the status of FDA’s foreign drug inspections and its use of alternative tools.[8] In this report, we
1. describe the status of FDA in-person drug inspections,
2. examine FDA inspection workforce vacancies and FDA’s efforts to address them,
3. describe FDA’s use of alternative tools during the COVID-19 pandemic and its plans for the future, and
4.
describe selected foreign regulators’ use of alternative tools to
oversee drug manufacturing establishments in other countries.
For all four objectives, we reviewed relevant federal laws, regulations, or other documentation related to oversight of drug manufacturing establishments. We also interviewed FDA officials from the Center for Drug Evaluation and Research (CDER), which identifies and prioritizes establishments for inspection, the Office of Regulatory Affairs (ORA), which is responsible for conducting the inspections, and the Office of Global Policy and Strategy, which oversees the activities of FDA’s foreign offices. In addition, we interviewed a nongeneralizable selection of 10 current investigators and former investigators who now have other roles at FDA (whom we refer to as “investigators” for the purposes of this report), and who conducted inspections and used alternative tools. We made this nongeneralizable selection to gather the perspectives of staff with a range of experiences with alternative tools, including experiences using more than one tool. The views of these investigators cannot be generalized to other investigators.
To describe the status of FDA in-person drug inspections, we analyzed FDA data on inspections of drug manufacturing establishments.[9] Specifically, we examined FDA data from fiscal year 2019 (the last full year of inspections prior to the start of the COVID-19 pandemic) through fiscal year 2023. Fiscal year 2023 data were the most recent year available when we conducted our analysis. To provide context for the number of inspections, we obtained data from FDA on the number of establishments the agency considered to be subject to inspection in each country as of January 2024, which was the most recently available data at the time of our analysis. Finally, we reviewed agency documents and interviewed agency officials about FDA’s efforts to resume in-person inspections following the COVID-19 pandemic.
To examine vacancies in FDA’s drug investigator workforce and the agency’s efforts to address them, we analyzed FDA data on the number of authorized, filled, vacant, newly hired, and departing investigator positions for fiscal years 2022 through June 30, 2024 (partial fiscal year 2024). Fiscal year 2022 was the first full fiscal year since we last reported on FDA’s efforts to address investigator vacancies; partial fiscal year 2024 was the most recently available data when we conducted our analysis. We also interviewed officials about their efforts to maintain a sufficient pool of drug investigators, and we reviewed FDA documents related to workforce planning and investigator recruitment and hiring. In addition, we interviewed investigators regarding their experiences conducting in-person inspections, including challenges related to inspection staffing. We compared FDA’s efforts and action plans to maintain its drug investigator workforce against selected leading practices for retention previously identified by GAO.[10]
To describe FDA’s use of alternative tools during the COVID-19 pandemic, and its future plans for these tools, we analyzed data from FDA’s Compliance Management System on the agency’s use of alternative tools from fiscal year 2021 (to update our prior analyses of use of such tools in fiscal year 2020) through fiscal year 2023 (the most recent complete year of data available at the time of our analysis). We also reviewed agency documents and interviewed agency officials—including FDA staff involved with setting policy about the past and future use of such tools—and investigators, about their experiences using these tools. Further, we reviewed documents from and interviewed six drug manufacturing industry stakeholder groups about the experiences of their member companies with FDA’s use of alternative tools.[11] These stakeholder groups represent the major components of the drug manufacturing industry: manufacturers of brand-name drugs, generic drugs, active ingredients, over-the-counter drugs, and contract manufacturers. Each of the stakeholder groups represents both member companies with domestic manufacturing establishments and those with foreign manufacturing establishments.
To describe selected foreign regulators’ use of alternative tools to oversee drug manufacturing establishments in other countries, we reviewed documents from and interviewed officials from seven foreign regulators. We selected regulators from Australia, Canada, Japan, New Zealand, Switzerland, the United Kingdom, and the European Union.[12] We selected these seven because they have inspection information sharing agreements with FDA, and they have documented use of alternative tools. We interviewed officials from the seven regulators about the types of alternative inspection tools they use and the challenges and benefits of using such tools.[13]
Our work focused on human drugs regulated by CDER.[14] Further, our work focused on activities related to FDA’s inspections of manufacturing establishments. FDA undertakes other activities to oversee drug quality, such as sampling and testing, which are beyond the scope of our review.[15]
To assess the reliability of the data on inspections, the number of establishments subject to inspection, investigator staffing, and the use of alternative tools, we reviewed related documentation, interviewed knowledgeable agency officials, conducted electronic data testing for missing data and outliers, and compared the data to published information from the same sources, as appropriate. On the basis of these steps, we found these data sufficiently reliable for the purposes of our reporting objectives.
We conducted this performance audit from April 2023 to November 2024 in accordance with generally accepted government auditing standards. Those standards require that we plan and perform the audit to obtain sufficient, appropriate evidence to provide a reasonable basis for our findings and conclusions based on our audit objectives. We believe that the evidence obtained provides a reasonable basis for our findings and conclusions based on our audit objectives.
Background
Globalization of Drug Manufacturing
Drugs sold in the U.S.—including active pharmaceutical ingredients and finished dosage forms—are manufactured throughout the world. These include brand-name, generic, and over-the-counter drugs. As of January 2024, FDA data showed that India and China had the most foreign establishments manufacturing drugs for the U.S. market, with nearly 40 percent of all foreign establishments in these two countries. (See fig. 1.)
Figure 1: Number of Establishments in the U.S. and the 10 Countries with the Most Foreign Drug Establishments Manufacturing Drugs for the U.S. Market, as of January 2024
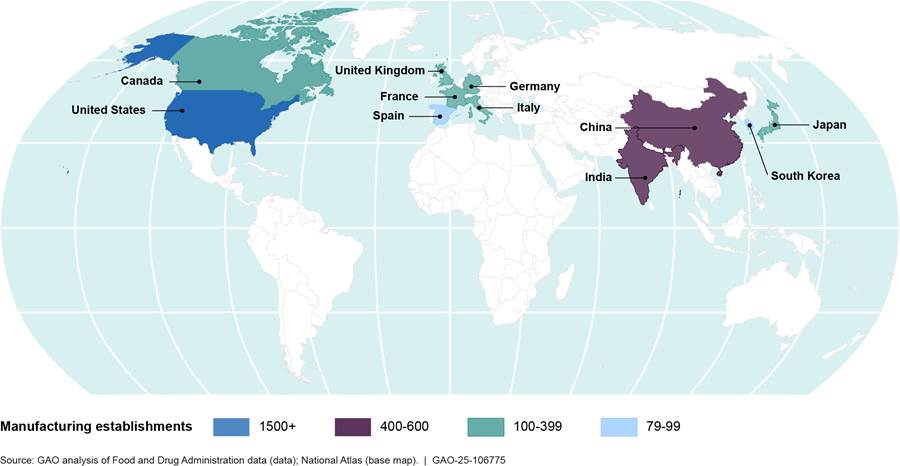
Note: This figure includes the 10 countries with the most foreign drug
establishments manufacturing drugs for the U.S. market and does not include
those countries with fewer than 79 establishments.
Types of Inspections
Drugs manufactured overseas for the U.S. market must meet the same statutory and regulatory requirements as those manufactured in the U.S. Under its applicable statutory and regulatory authority, FDA’s CDER implements requirements for the safety, quality, effectiveness of, and manufacturing processes for, over-the-counter and prescription drugs. CDER requests that ORA inspect both domestic and foreign establishments to ensure that drugs are produced in conformance with applicable laws of the U.S., including FDA regulations governing current good manufacturing practices (GMP).[16] ORA investigators conduct the inspections.
Investigators generally conduct three main types of
in-person drug manufacturing establishment inspections: preapproval
inspections, surveillance inspections, and for-cause inspections, as described
in
table 1.[17]
|
Type of inspection |
Purpose of inspection |
|
Preapproval inspections |
FDA conducts preapproval inspections before approving a new brand-name or generic drug to be marketed in the U.S. These inspections are designed to verify the accuracy and authenticity of drug application data (such as manufacturing records) to determine that the establishment is following commitments made in the application and to assess whether the establishment can manufacture the product in the application in conformance with applicable regulations to ensure a drug’s identity, strength, quality, and purity.a |
|
Surveillance inspections |
Surveillance inspections are conducted at establishments when drugs are already marketed in the U.S.—either after FDA approval or after marketing for drugs that do not require FDA approval before marketing—and focus on compliance with system-wide controls for ensuring that the manufacturing processes produce high-quality drugs.b Systems examined during these inspections include those related to materials, quality control, production, facilities and equipment, packaging and labeling, and laboratory controls. These systems may be involved in the manufacture of multiple drugs. |
|
For-cause inspections |
For-cause inspections are conducted to investigate specific issues, such as those raised in consumer complaints, reports of product quality issues submitted by consumers or health care professionals, indications of potential manufacturing problems submitted by the manufacturers themselves, or to follow-up on previous FDA regulatory action, among other reasons. |
Source: GAO analysis of Food and Drug Administration (FDA) information. │ GAO‑25‑106775
aWhen FDA receives an application for drug approval (or a supplement to that application related to a manufacturing change), officials review the inspection history of each establishment listed on the application, among other things. According to FDA officials, if an establishment listed on the application has received a satisfactory good manufacturing practices inspection for a similar or more complex product, and the agency has no new concerns, FDA may consider this inspection sufficient and not perform a preapproval inspection of this establishment. FDA may also conduct post-approval inspections that focus on a specific product and are conducted after applications have been approved. Post-approval inspections largely focus on the process validation lifecycle and any manufacturing changes that may have occurred following approval.
bCertain drugs, such as some over-the-counter
drugs, may not require FDA approval before being marketed in the U.S.
While preapproval and for-cause inspections occur in response to specific needs, FDA uses a risk-based process to select establishments for surveillance inspections.[18] CDER first compiles a catalog of the more than 4,000 establishments that are subject to inspection. CDER then applies a risk-based site selection model to the catalog to prioritize establishments for surveillance inspection. Using the results of the model and other information, CDER develops a ranked list of foreign and domestic establishments that FDA considers to be a priority for inspection in a given year and submits that list to ORA. To determine how many inspections to request that ORA conduct in a given year, CDER receives information from ORA on available inspection resources and determines how these resources should be budgeted across multiple inspection programs and other activities, according to FDA officials. Thus, not every establishment will be inspected each year. For example, in fiscal year 2019, there were about 4,200 establishments in FDA’s catalog of establishments subject to inspection and FDA conducted about 1,700 inspections.
FDA Inspection Workforce
FDA has three groups of investigators who conduct domestic and foreign drug manufacturing inspections.
· General pool of investigators. Investigators based in the U.S., who primarily conduct domestic inspections, but who also conduct foreign inspections.
· Dedicated foreign drug cadre. A U.S.-based group of investigators who specialize in foreign inspections.
· Foreign office investigators. Investigators based in FDA’s India or China offices.
We previously identified persistent vacancies among these three groups of investigators. We recommended that FDA develop strategies focused on the recruitment and retention of investigators who specialize in foreign inspections.[19] FDA agreed with our recommendation and in response, in December 2021 FDA formed the GAO Recruitment and Retention Action Plan Work Group, which developed six tailored strategies to recruit, develop, and retain investigators for the foreign drug cadre and the foreign offices.[20] For example, FDA participated in recruiting events and outreach to individuals with interest in or skills relevant to travel, such as former Peace Corps volunteers. FDA also increased the cash incentive for completed foreign trips and launched a three-part training series on foreign travel. As a result of these efforts, we determined that FDA had partially implemented our recommendation. As of July 2024, FDA stated that it planned to form a work group to address recruitment challenges specific to the foreign offices but had not done so. However, FDA stated that ORA and the Office of Global Policy and Strategy continued to work together to address challenges related to the recruitment and retention of foreign office investigators. To fully meet the intent of our recommendation, FDA should detail implementation steps and time frames for its proposed efforts related to foreign office recruitment.
Alternative Tools
While FDA has historically relied on in-person inspections to oversee domestic and foreign establishments, it can also utilize multiple types of alternative tools, in certain circumstances. These generally fall into two categories: review of reports from inspections conducted by foreign regulators, and the remote assessment of information provided by establishments. (See fig. 2.)

Foreign regulator inspection reports. FDA has agreements with regulators
around the world that allow it to share information about drug establishments.
In 2012, federal law authorized FDA to enter into mutual recognition agreements
to recognize inspections conducted by foreign regulators deemed capable of
conducting inspections that meet U.S. requirements.[21] Since then, FDA has entered into
mutual recognition agreements with European regulators to use each other’s
inspection reports, which can reduce duplicative inspections.[22] In contrast, FDA has confidentiality
commitments with certain regulators, which allow FDA to share certain kinds of
inspection information but do not establish that each regulator will recognize
the other’s inspections.
In March 2020, FDA expanded its use of reports obtained through mutual recognition agreements and those obtained through other sharing agreements.[23] First, FDA began recognizing inspections that European regulators conducted outside of Europe, such as in China and India.[24] Second, FDA began expanding use of inspection reports from regulators that are among the members of the Pharmaceutical Inspection Co-operation Scheme, which includes 43 regulators with which FDA shares another agreement and 13 regulators with which FDA does not have an existing agreement, according to FDA.[25] Rather than substituting a report for an FDA inspection, as it can do under mutual recognition agreements, FDA used these reports from other regulators in conjunction with information obtained by FDA through other methods to make oversight decisions.
Remote assessments. FDA may also conduct an examination of an establishment or its records entirely remotely to evaluate compliance with applicable FDA requirements. Remote assessments include the following activities.
· Remote records reviews. In 2012, federal law authorized FDA to request and review records and other information from drug manufacturing establishments subject to inspection.[26] FDA may request records it would typically review during a preapproval or surveillance inspection or may make more targeted requests. Prior to the COVID-19 pandemic, FDA used this authority in a more limited capacity, for example to oversee 10 establishments that the agency would not routinely inspect because of travel warnings.[27]
· Remote interactive evaluations. In April 2021 guidance, FDA described various remote interactive tools the agency may use to conduct an evaluation of an establishment during the COVID-19 public health emergency.[28] Such tools included use of teleconference, livestream video, and screen-sharing of data and documents.
FDA Resumed Inspections Paused during the COVID-19
Pandemic and Progressed with Two In-Person Inspection Pilot Programs
By the start of fiscal year 2023, FDA had resumed conducting in-person inspections paused during the pandemic, but inspection totals remained below pre-pandemic totals due to reduced investigator capacity. As a part of resuming in-person inspections, FDA implemented the unannounced inspections pilot and finalized the design of the independent interpreter pilot.
By Fiscal Year 2023, FDA Had Resumed Inspections Paused during Pandemic Disruptions but Number of Inspections Had Not Returned to Pre-Pandemic Inspection Levels
In fiscal year 2023, FDA conducted more drug inspections than it had since pausing most inspections in March 2020, at the start of the COVID-19 pandemic, but did not reach pre-pandemic inspection totals, according to our analysis of FDA data.[29] (See fig. 3.) These data show FDA conducted 1,065 total inspections in fiscal year 2023, a 40 percent increase from fiscal year 2022. However, this total was 36 percent below the 1,671 inspections conducted in fiscal year 2019, the last full year before the start of the pandemic.
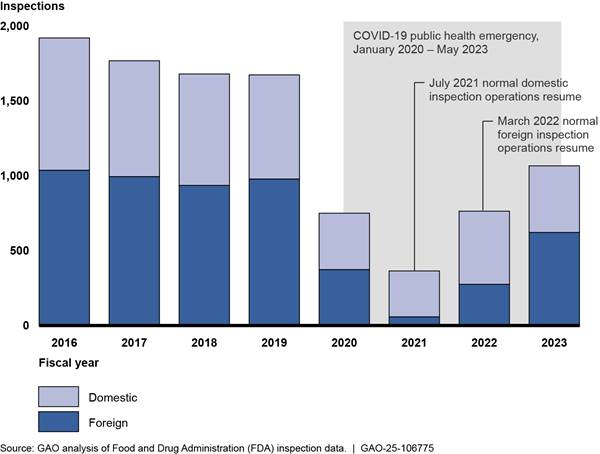
Note: FDA paused all inspections except those deemed mission critical in March 2020 due to the COVID-19 pandemic. The agency resumed normal operations for domestic inspections in July 2021 and began resuming normal operations for foreign inspections in March 2022, according to FDA officials.
The number of inspections FDA conducted was limited by reduced investigator capacity, according to FDA officials. FDA officials told us that they anticipate that fiscal year 2024 inspection totals will be higher than fiscal year 2023 but will remain below pre-pandemic levels due to continued reduced investigator capacity.[30]
As FDA has resumed conducting inspections, surveillance inspections have increased and trends in inspection location and outcomes have become more similar to what FDA conducted prior to the COVID-19 pandemic.
Inspection location. Our analysis of FDA data found that FDA conducted mostly domestic inspections following the start of the pandemic in 2020. However, by fiscal year 2023, 58 percent of drug inspections were of foreign establishments, which was the same as the percent of inspections that were foreign in fiscal year 2019, prior to the pandemic. Similarly, in fiscal year 2023, FDA again conducted the largest number of foreign inspections in India and China, where nearly 40 percent of foreign establishments are located.[31] (See table 2.)
|
Country |
2019 |
2020 |
2021 |
2022 |
2023 |
Number of establishments subject to |
|
India |
305 |
155 |
9 |
83 |
212 |
592 |
|
China |
167 |
30 |
25 |
17 |
90 |
487 |
|
Switzerland |
29 |
10 |
1 |
9 |
37 |
77 |
|
Canada |
70 |
28 |
0 |
30 |
33 |
130 |
|
Japan |
51 |
33 |
8 |
5 |
32 |
124 |
|
Germany |
69 |
12 |
1 |
14 |
20 |
195 |
|
All other foreign countries |
286 |
106 |
15 |
118 |
197 |
1,158 |
|
Foreign (total) |
977 |
374 |
59 |
276 |
621 |
2,763 |
|
Foreign (percent of total inspections) |
58% |
50% |
16% |
36% |
58% |
58% |
|
Domestic (total) |
694 |
376 |
306 |
487 |
444 |
1,980 |
|
Domestic (percent of total inspections) |
42% |
50% |
84% |
64% |
42% |
42% |
|
Total |
1,671 |
750 |
365 |
763 |
1,065 |
4,743 |
Source: GAO analysis of Food and Drug Administration (FDA)
data. | GAO‑25‑106775
Note: Drug manufacturing inspection counts include FDA’s inspections of establishments as part of its drug approval process, to conduct regular surveillance after drugs are marketed in the U.S., and to investigate specific issues. Inspection counts do not include those inspections completed using inspection reports from foreign regulators with which FDA has a mutual recognition agreement. FDA can substitute these reports for its own inspections. Of the countries included in this table, FDA has had a mutual recognition agreement with Germany since 2017 and with Switzerland since 2023.
aJanuary 2024 was the most current data
available on the number of establishments subject to inspection at the time we
conducted our analysis. The number of establishments subject to inspection in a
given country can vary over time. However, the overall trend in the proportion
of foreign to domestic sites has generally remained the same over the past
several fiscal years. (For an analysis of changes in the number of
establishments subject to inspection in the above countries between fiscal year
2019 and 2023, see U.S. Food & Drug Administration, Center for Drug
Evaluation and Research, Fiscal Year 2023 Report on the State of
Pharmaceutical Quality (June 2024).)
|
Surveillance Backlog In 2021, we reported that the postponement of surveillance inspections due to the COVID-19 pandemic created an inspection backlog that could affect the Food and Drug Administration’s (FDA) goal of conducting exclusively risk-driven surveillance inspections. Specifically, as a result of postponements, the number of establishments never inspected or not inspected in the past 5 years increased. This left fewer inspection resources for other establishments identified by FDA’s risk-based site selection model as having the greatest potential for public health risk if the site is out of compliance. We recommended that FDA ensure its future inspection plans analyze and respond to the issues presented by this backlog of surveillance inspections.
FDA agreed with our recommendation and has implemented it. For example, in fiscal year 2024, FDA planned to use abbreviated inspections of certain low risk establishments to increase the total number of surveillance inspections it can complete. As a result, in fiscal year 2024, the proportion of planned surveillance inspections that are of relatively higher risk establishments has increased for the first time since the start of the COVID-19 pandemic, in alignment with FDA’s goal of risk-driven surveillance. However, the backlog caused by postponements continues to be a factor in FDA’s selection of establishments to inspect. Source: GAO review of FDA inspection plans (text); aerogondo/stock.adobe.com (photo). | GAO-25-106775 |

Inspection type. During the COVID-19 pandemic pause in most inspections, FDA considered certain preapproval and for-cause inspections to be mission critical or otherwise of higher priority than routine surveillance inspections.[32] Thus, of the limited number of inspections conducted during fiscal year 2021, the first full fiscal year affected by the pandemic, FDA conducted more for-cause inspections than surveillance inspections. (See sidebar for more details on the postponement of surveillance inspections.) In fiscal years 2022 and 2023, surveillance inspections were the most common type of inspection, according to our analysis.
However, FDA documents and officials note that for-cause inspections will likely continue to exceed pre-pandemic levels. This is due to multiple factors, according to FDA officials. For example, for-cause inspections are necessary to follow-up on compliance actions, such as import alerts, taken as a result of targeted remote records reviews conducted during the pandemic. FDA also noted a general increase in the receipt of information that can trigger a for-cause inspection.
|
Inspection Classifications Based on inspection findings and the Food and Drug Administration’s (FDA) review, each inspection is classified into one of the following categories based on FDA’s determination of whether any deficiencies identified during the inspection are serious enough to warrant regulatory action: No action indicated means that insignificant or no deficiencies were identified during the inspection. Voluntary action indicated means that deficiencies were identified during the inspection, but the agency is not prepared to take regulatory action, so any corrective actions are left to the establishment to take voluntarily. Official action indicated means that serious deficiencies were found that warrant regulatory action. For example, FDA identified possible cross contamination during an inspection, which led to a recall of multiple products.
|
Inspection outcomes. In fiscal years 2021 and 2022, during the COVID-19 pandemic, a higher percentage of inspections FDA conducted received FDA’s most serious classification of official action indicated, as compared to before the pandemic. (See sidebar.) For example, in fiscal year 2021, 30 percent of inspections received an official action indicated classification compared to 13 percent in fiscal year 2019. However, the total number of inspections that received an official action indicated classification did not increase. (See fig. 4.) This higher percentage may reflect the fact that for-cause inspections have increased in recent years, according to FDA documents, which officials noted are more likely to have deficiencies. In fiscal year 2023, as routine surveillance inspections increased, the percentage of inspections classified as official action indicated decreased and was similar to pre-pandemic rates.[33]
Figure 4: Number and
Percentage of FDA Foreign and Domestic Inspection Classifications, Fiscal Years
2019–2023
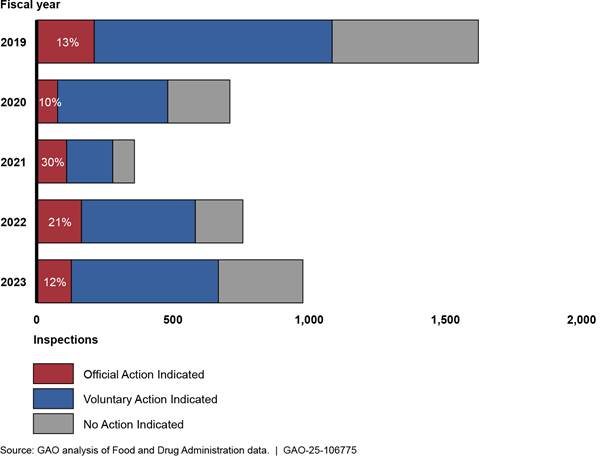
Note: After each inspection, FDA classifies the inspection into one of three categories based on its determination of whether any deficiencies identified during the inspection are serious enough to warrant regulatory action: No Action Indicated means that insignificant or no deficiencies were identified during the inspection; Voluntary Action Indicated means that deficiencies were identified during the inspection, but the agency is not prepared to take regulatory action, so any corrective actions are left to the establishment to take voluntarily; and Official Action Indicated means that serious deficiencies were found that warrant regulatory action.
Inspection totals do not sum to 100 percent due to
rounding and because some inspections had not yet received a final
classification as of the date that FDA pulled these classification data.
FDA Implemented Its Unannounced Inspections Pilot and Has Finalized the Design of Its Independent Interpreter Pilot
As it resumed inspections paused by the COVID-19 pandemic, FDA implemented one pilot program and finalized the design of another pilot program that are both intended to address unique challenges to conducting foreign inspections. They are (1) the unannounced inspections pilot and (2) the independent interpreter pilot. Historically, foreign inspections were generally preannounced, and investigators generally relied on the establishment being inspected to provide translation services. These unique challenges raised questions about the equivalence of foreign to domestic inspections.[34]
Unannounced inspections pilot. FDA began implementation of this pilot program in March 2022. Consistent with our recommendation, FDA incorporated all five of our leading practices into the pilot’s design and implementation. For example, consistent with leading practices, FDA developed a pilot evaluation plan that outlines an assessment methodology and a plan for collecting both quantitative and qualitative data to assess the effect of conducting unannounced versus preannounced inspections across a range of metrics.[35] Such data include information from FDA data systems on the time and financial resources spent planning and conducting inspections, surveys of investigators that participated in the pilot, and documentation of observations made during the inspections.
FDA is implementing this pilot in India and China in three phases, as outlined in figure 5.
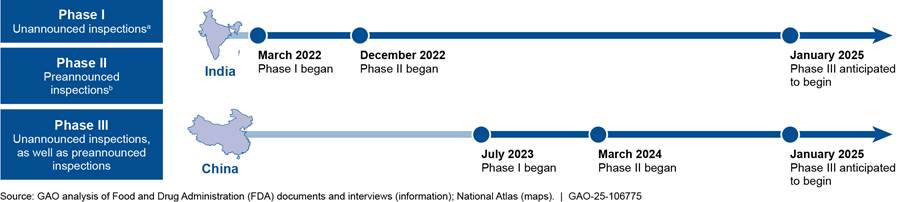
aUnannounced inspections include those announced on short notice—those in which establishments receive notice of the inspection 72 hours or less before the inspection begins. Establishments may receive much less than 72 hours’ notice, including less than 1 hour.
bPreannounced inspections are announced to the
establishment 8 or more weeks in advance of the inspection.
According to FDA officials, pilot implementation in China was slowed by COVID-19-related travel restrictions, a new visa application process, and new Chinese laws related to espionage and national security. In addition, FDA officials said that increased resource needs for pilot inspections have affected the pace of implementation. Specifically, FDA determined that unannounced inspections in the pilot are to be conducted by two investigators for safety reasons (historically, the majority of inspections were conducted by solo investigators).[36]
As of May 2024, FDA had initiated 114 pilot inspections in India (94 of which were unannounced) and 28 in China (16 of which were unannounced), according to an FDA presentation on the pilot’s status. FDA plans to continue pilot implementation through each phase until it has completed about 250 unannounced and about 250 preannounced inspections in total across both countries.
FDA plans to conduct evaluations at the end of each phase in each country and does not expect to complete the final evaluation until all phases are complete in both countries, which FDA officials anticipate will take several years. FDA completed an internal evaluation of the first phase of the India pilot in April 2023 and gained a number of insights. For example, of the unannounced inspections initiated in India during the first phase of the pilot, FDA noted the following.
· There were two instances in which FDA discovered that a regulatory agency from another country was already on site when FDA arrived at the establishment. According to the evaluation, in these cases FDA and the establishment did not encounter any challenges with this overlap.
· Two inspections could not be completed as planned. Upon initiating the inspection, FDA determined that one establishment was out of business. The second establishment was on import alert and establishment staff told FDA investigators that the establishment was not ready for inspection.[37] In both instances, investigators were re-assigned to an unannounced inspection at another establishment the next day.
According to FDA officials, in both instances FDA learned that these potential challenges could be overcome, and the pilot did not need to be redesigned.
Independent interpreter pilot. Our review of FDA’s April 2023 pilot design document shows that, consistent with our recommendation, FDA incorporated all five leading practices into its design; for example, FDA established two measurable pilot objectives. For the first objective, FDA plans to assess whether the source of an interpreter (either hired by FDA or provided by the inspected establishment) has an effect on an investigator’s ability to conduct a comprehensive and timely inspection. For the second, FDA plans to assess the costs, resources, benefits, and challenges of each interpreter source.
According to pilot design documentation, FDA plans to conduct this pilot in mainland China using interpreters from the State Department. FDA plans to conduct between 34 and 119 inspections as part of the pilot, with the exact number to be determined based on the level of precision desired for the pilot evaluation. Pilot inspections will include preapproval, surveillance, and for-cause inspections to control for variance between inspection types.
FDA reported in May 2024 that implementation of the interpreter pilot was not likely to begin until after fiscal year 2024. FDA reported that it was prioritizing inspection resources for the unannounced inspections pilot and continuing to assess the evolving landscape in China to determine the most appropriate time to launch this pilot. To fully implement our recommendation, FDA needs to provide documentation of pilot implementation.
FDA Is Taking Action to Address Investigator Vacancies but Does Not Have Plans That Address All Root Causes of Attrition
Investigator attrition has led to an increase in drug
investigator vacancies since November 2021. According to FDA, these vacancies,
along with the resulting large number of relatively inexperienced
investigators, has limited the number of drug inspections FDA can complete. FDA
has identified the root causes of investigator attrition and has some
strategies underway to address them, but the agency has not developed action
plans that address all the identified root causes, due in large part to the
fact that implementing such plans would reduce capacity to conduct inspections.
Investigator Attrition Led to Increased Vacancies, Which FDA Says Limited the Number of Inspections Conducted
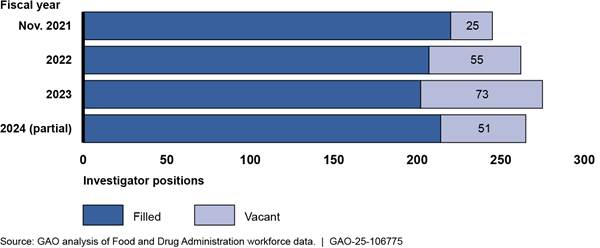
FDA data show the total number of investigator vacancies has increased from 25 vacant positions in November 2021 to 51 as of June 2024.[38] Across the three groups of investigators, the largest increase in vacancies was in the general pool of investigators who conduct both domestic and foreign inspections. However, vacancies also persist in the dedicated foreign drug cadre and among investigators in FDA’s foreign offices. (See sidebar for information on FDA’s three groups of investigators.)
|
Foreign Inspections by Investigator Type The Food and Drug Administration (FDA) relies on a drug inspection workforce primarily composed of three groups of investigators based both in the U.S. and overseas. · General pool of investigators. Investigators based in the U.S., who primarily conduct domestic inspections, but who also conduct foreign inspections. This is the largest group of investigators, and therefore, conduct the majority of foreign inspections. They conducted about 72 percent of all foreign inspections in 2019. · Dedicated foreign drug cadre. A U.S.-based group of investigators who specialize in foreign inspections. They conducted about 18 percent of all foreign inspections in 2019. · Investigators in foreign offices. Investigators based in FDA’s India or China offices. They conducted about 11 percent of all foreign inspections in 2019. All investigators begin their careers in the general pool based in the U.S., conducting only domestic inspections. Investigators may become eligible to conduct foreign inspections, join the cadre, or move to a foreign office after gaining experience and training. Source: GAO review of FDA information. | GAO‑25‑106775 |
General pool of investigators based in the U.S. Most of the current drug investigator vacancies are in the general pool of investigators, which is the largest group of investigators and conducts the majority of domestic and foreign inspections. There are about 230 total authorized investigator positions in this pool. We previously reported that, from December 2019 to November 2021, FDA made progress in hiring for the general pool of investigators.[39] According to FDA, in November 2021, there were 20 vacancies in this pool (a 9 percent vacancy rate).[40] However, as of June 2024, the number of vacancies had increased to 37, a 16 percent vacancy rate. (See fig. 6.)
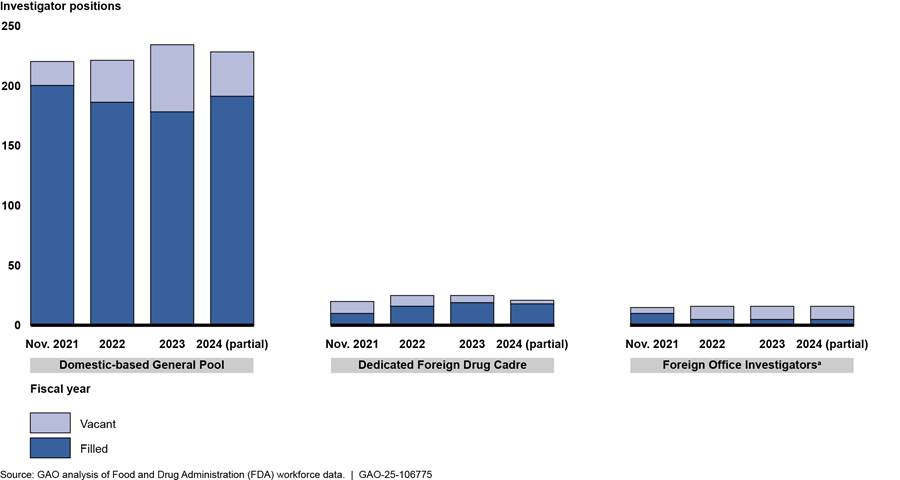
Note: In our prior report, we counted certain investigator positions as filled for which a new hire had a scheduled start date but had not yet begun work. (See GAO, Drug Safety: FDA Should Take Additional Steps to Improve Its Foreign Inspection Program, GAO‑22‑103611 (Washington, D.C., Jan. 7, 2022).) The data FDA provided for this report, including revised data for November 2021, include only those investigators officially onboard as of that date. Additionally, since our prior report, FDA has determined that certain positions which only occasionally conduct inspections should be excluded from the count of onboard investigators that FDA provided for the previous report.
aThis represents a combined vacancy rate for India and China, the two foreign office locations where FDA has drug investigator positions. Vacancies are greater in China than in India. For example, as of June 2024, four of seven positions in India were vacant, a 57 percent vacancy rate, and seven of nine positions in China were vacant, a 78 percent vacancy rate.
Dedicated foreign drug cadre investigators. FDA recruited more investigators into the dedicated foreign cadre and expanded the number of possible positions, though vacancies remain. As of June 2024, 18 of the 21 cadre positions were filled, compared to 10 of 20 positions as of November 2021.[41] Although FDA has not yet filled every vacancy, according to officials, FDA increased the total number of positions in the cadre because of funding received to conduct inspections under the unannounced inspections pilot, which were expected to largely be conducted by cadre investigators.
Foreign office investigators. Vacancies increased among these investigators, particularly in China.[42] FDA data show that, in November 2021, there was one vacancy among six positions in India and four vacancies among nine positions in China. As of June 2024, four of seven positions in India were vacant and seven of nine positions in China.[43] (See text box for examples of challenges filling positions in China.)
|
Challenges Specific to Filling Positions in FDA’s Foreign Office in China Food and Drug Administration (FDA) officials noted several challenges specific to filling positions in the China office, including restrictions during the COVID-19 pandemic and concerns around the invasiveness of national laws. According to officials, China’s strict zero-COVID policy and related requirements during much of the COVID-19 pandemic resulted in three staff ending their China assignments early. Post-pandemic, officials stated that staff remain concerned about the potential for future lockdowns and raised concerns around the arbitrary enforcement of local and national security laws. For a previous report, officials told us that the challenge of recruiting for positions in China is not unique to FDA, as other agencies with a presence in China also have vacancies. In addition, as of April 2024, the State Department had issued a Level 3 Travel Advisory for China (avoid travel due to serious risks to safety and security). |
Source: GAO analysis of Food and Drug Administration
information. | GAO‑25‑106775
FDA has continued to recruit and hire investigators, but vacancies persist due to challenges with investigator attrition. According to FDA data, from the start of fiscal year 2022 to June 2024, ORA hired 105 new investigators into the general pool of approximately 230 authorized investigators and lost 105. Of these 105 drug investigator losses, 61 percent left for other FDA positions, including other positions in ORA; 29 percent left FDA; and 10 percent retired.[44] The turnover rate for ORA investigators in the general pool was more than twice as high as the ORA-wide turnover rate in fiscal years 2022 and 2023. (See table 3.) As of June 2024, hiring for fiscal year 2024 has been greater than attrition. However, officials noted that they expected additional attrition, as investigator turnover has been an ongoing issue.
|
|
Fiscal year 2022 |
Fiscal year 2023 |
Partial fiscal year 2024 (as of June) |
|
Investigator gains |
35 |
35 |
35 |
|
Attrition |
41a |
40 |
24 |
|
Net change |
-6 |
-5 |
+11 |
|
Turnover rate |
22% |
22% |
13% |
Source: GAO review of Food and Drug Administration (FDA) data. │ GAO‑25‑106775
aThis includes two investigators who accepted
assignments to a foreign office. During the time that investigators from FDA’s
Office of Regulatory Affairs are posted to a foreign office, they are detailed
to the Office of Global Policy and Strategy, a different office within FDA.
High rates of attrition in the general pool of investigators limit FDA’s ability to fill positions in the foreign cadre and the foreign offices, which are filled by more experienced investigators from the general pool.[45] Officials told us that new investigators typically need 2 to 3 years of experience before they can conduct foreign inspections independently, and they must have experience conducting foreign inspections before they are qualified for a position in the foreign cadre or for an assignment as a foreign office investigator. As of June 2024, 36 percent of investigators in the general pool did not have the necessary experience to conduct independent foreign inspections. Officials told us that the limited pool of qualified investigators was one of the top challenges in recruiting investigators to fill foreign office investigator positions.
Investigator attrition has reduced the total number of inspections FDA can conduct, according to FDA documents and officials. Having a less-experienced investigator workforce has also reduced inspection capacity. FDA officials noted that newly hired investigators do not have the same capacity to complete inspections as experienced investigators. In addition, experienced investigators are less efficient in completing inspections because they are training newer staff. According to our review of FDA data and an FDA document, about 63 percent of current investigators were hired within the last 5 years, as of May 2024. Officials stated that, even if the total number of drug investigators currently on board was similar to past years, it is an enormous challenge to maintain a similar number of inspections to previous years with so many inexperienced investigators.
FDA Identified Root Causes of Attrition and Has Taken Some Steps to Address Them but Does Not Have Action Plans for All Causes
ORA, which hires and manages investigators, led FDA efforts to identify the root causes of attrition. ORA is implementing action plans to address some but not all of these root causes. Fully addressing all root causes may require agency-wide collaboration.
FDA identified attrition root causes. Based on our review of ORA documentation and FDA interviews, we found that ORA identified travel, pay, training, workload, and work-life balance concerns as root causes of investigator morale issues leading to attrition. According to officials, ORA initially identified these root causes of attrition through its GAO Recruitment and Retention Action Plan Work Group, which was formed in fiscal year 2022 in response to our prior recommendation.[46] ORA officials stated that they reviewed employee exit surveys and attrition data, as well as data from the annual Federal Employee Viewpoint Survey and employee focus groups.[47] Officials stated that they continue to review these sources on an ongoing basis. Based on their review, officials noted travel and pay as the main causes of attrition. FDA investigators we interviewed also described challenges that align with these root causes.[48]
· Travel. Officials identified travel as the biggest cause of attrition, including both the amount of travel and the conditions of travel. Investigators travel frequently, from 25 to 75 percent of the time according to position descriptions, and may spend long hours in transit, according to officials. Officials provided an example of a trip to China where the investigator left home on Thursday and spent 41 hours in transit to arrive for a Monday inspection.[49] Five investigators we interviewed identified travel as a challenge, including the amount of time away from home and concerns with safety on foreign inspection trips, which are often conducted by a single investigator.[50] One investigator described being assaulted on a foreign trip. Although officials stated foreign travel was the largest concern, they noted that domestic inspections also require a lot of travel and time away from home.
· Pay. According to officials, departing drug investigators often cite better job opportunities, including increased pay, as a reason for leaving the investigator position. FDA workforce data show that the majority of investigators who leave transfer to other positions in FDA where, according to officials, they receive the same or higher pay with less required travel. For example, of those that transferred to other FDA centers from October 2021 through February 2024, at least 29 percent received a salary increase.[51] The average salary increase was 30 percent. However, at least 42 percent of those transferring to other FDA centers accepted a position with an equivalent salary, indicating that increased pay is not always the cause of attrition.
· Training. Ensuring sufficient and standardized training for new investigators was identified as a concern by employee focus group participants and in some exit interviews. For example, focus group participants wanted more hands-on training and tools to standardize the on-the-job training.
· Workload. Officials stated that investigators have a heavy workload during an inspection and a short time frame to complete their inspection reports when they return from a trip. For example, an investigator on a three-week foreign trip covering three different inspections may have their first inspection report due before they return. Four investigators stated that they frequently work overtime during inspections and to complete inspection reports and other tasks.[52]
· Work–life balance. Officials noted that it is difficult for investigators to maintain a work-life balance and make personal commitments, such as being present for child or family events, when they travel so frequently. As a result, officials said investigators take other jobs that are less stressful and require less travel, or that offer remote work options. In FDA exit interviews, departing employees noted that workload expectations made it difficult to have a healthy personal life
and also cited an unreasonable amount of unplanned travel. For example, one investigator noted that investigators may receive a 2-day notice to travel for a domestic inspection.
Changing internal and external factors since the pandemic have further compounded existing issues of travel, workload, and work-life balance. For example, FDA’s increased use of inspection reports from European regulators has reduced the need to conduct inspections in Europe, according to officials. As a result, a larger proportion of foreign trips are to India and China, increasing the total travel time and travel hardship for investigators. Additionally, officials noted that since the pandemic many non-investigator jobs offer telework or remote work opportunities, which may offer better work-life balance.
These root causes of attrition are not always distinct. In particular, issues of travel, workload, and work-life balance are often related and lead to investigator burnout according to officials. For example, some work-life balance issues are the result of travel schedules and workload from internal requirements.
|
Hiring and Pay Flexibilities Most of the Food and Drug Administration’s (FDA) workforce is hired under authority provided by Title 5 of the U.S. Code. The Title 5 hiring process, known as competitive examining, requires agencies to take specific steps related to public notice; screen applications against minimum qualification standards; apply selection priorities such as veterans’ preference; and assess all candidates against job-related criteria. Individuals hired under Title 5 in professional, technical, administrative, and clerical positions typically have their rates of pay assigned under the General Schedule classification system. Hiring flexibilities refer to hiring conducted outside of the competitive examining process, which may include authority under a different title of the U.S. code for the hiring of specific positions or to meet specific or critical needs. Hiring flexibilities may allow agencies to simplify the hiring process. The 21st Century Cures Act provided FDA with special pay authorities that provide for greater flexibility in setting pay, such as through the establishment of alternative pay bands with a higher maximum annual pay than is available under the General Schedule. Under this authority, FDA may convert or reappoint qualified existing staff to these alternative pay bands. Source: GAO review of
federal hiring authorities. | |
Action plans implemented. ORA has developed and is implementing action plans addressing the root causes of pay and training. According to ORA officials, they prioritized these as they were within ORA’s control and could be addressed using existing human capital guidance and flexibilities.
· Pay. ORA is expanding the use of Title 21 hiring authorities to include all investigators, which allows them to offer a higher maximum salary than is available under the traditional General Schedule pay system.[53] (See sidebar and table 4.) The use of Title 21 hiring and pay authorities was previously focused on certain FDA centers (such as CDER) and positions that support the development, review, and regulation of medical products. As a result, some investigators previously left ORA to take a Title 21 position at an FDA center, according to FDA salary data and officials. ORA began its expansion of Title 21 hiring authorities in fiscal year 2022 by converting certain investigator positions at particular risk of attrition to Title 21 positions, starting with the foreign cadre positions. In fiscal year 2024, ORA began hiring most new investigators under Title 21, according to officials, and will next focus on converting existing investigators to Title 21, according to an agency document.[54]
Table 4: Examples of Higher Maximum Pay Ranges for FDA Drug Investigators under Title 21 Pay Bands Compared to the General Schedule Pay Scale for the Washington, D.C., Region, as of January 2024
|
Type of staff and pay bands |
Minimum annual pay |
Maximum annual pay |
|
Associate Investigators |
|
|
|
General Schedule (GS-7)a |
$55,924 |
$72,703 |
|
Title 21 (pay band W)b |
$55,924 |
$74,155 |
|
Investigators I and II |
|
|
|
General Schedule (GS- 9-11) |
$68,405 |
$107,590 |
|
Title 21 (pay bands Y or A) |
$68,405 |
$109,506 |
|
Senior Investigators I |
|
|
|
General Schedule (GS-12) |
$99,200 |
$128,956 |
|
Title 21 (pay band B) |
$99,200 |
$133,845 |
|
Senior Investigators IIc |
|
|
|
General Schedule (GS-13) |
$117,962 |
$153,354 |
|
Title 21 (pay band C) |
$117,962 |
$164,260 |
|
Senior Investigators III |
|
|
|
General Schedule (GS-14) |
$139,395 |
$181,216 |
|
Title 21 (pay band D) |
$139,395 |
$191,900 |
Source: GAO analysis of documentation from Food and Drug Administration (FDA) and from the Office of Personnel Management. | GAO‑25‑106775
a5 U.S.C. ch. 31, subch. I. The General Schedule pay system typically applies to those hired under Title 5 of the U.S. code.
b21 U.S.C. 379d-3a. Hiring authority under Title 21 was originally provided by the 21st Century Cures Act. Pub. L. No. 114-255, 3072(a), 130 Stat. 1033, 1134 (2016). The Food and Drug Omnibus Reform Act of 2022 expanded this authority to apply to more positions and offices within FDA. Pub. L. No. 117-328, 3624, 136 Stat. 5807, 5879.
cAccording to an FDA document, external applicants are eligible for Investigator I and II and Senior Investigator I positions, depending on education and experience. Senior Investigator II positions and higher require existing FDA experience.
· Training. ORA expanded on-the-job training opportunities in February 2022 to include voluntary early participation in foreign inspections along with a senior investigator and also created working groups to oversee training proposals. ORA’s annual operational plans continue to include various training improvements, standardization efforts, and evaluations.[55] These efforts are intended to ensure newly hired investigators can independently conduct domestic inspections within 18 months and ensure current staff stay informed on industry and technology changes, according to ORA’s operational plans.
Unaddressed root causes. ORA, and FDA overall, have not implemented action plans to fully address three root causes of investigator attrition the agency has identified: travel, workload, and concerns with work-life balance. Leading practices for employee retention identified by GAO state that agencies should determine root causes of employee morale problems that lead to attrition and take action to address them by developing and implementing action plans linked to root causes.[56] According to the Office of Personnel Management, developing a workforce action plan involves the identification of strategies, plans to implement these strategies, and measures for assessing progress.[57] Furthermore, the Office of Personnel Management’s steps for action planning state that agencies should identify the necessary budget, resources, staff, and approvals needed to take action.
According to ORA officials, they have discussed options that may help address the amount of travel, investigator workload, and work-life balance issues, but they have not developed action plans because implementing such plans would reduce ORA’s current capacity to conduct inspections. ORA would thus not meet inspection goals set by CDER. For example, ORA discussed options to temporarily move some investigators to other positions that do not require travel or reduce the total number of weeks of foreign travel investigators are expected to complete in a year. However, ORA has not gone beyond dialogue to develop action plans. This is because of the potential effect on current inspection capacity that would be caused by reducing the number of inspections or weeks of foreign travel conducted by individual investigators.
For certain travel and workload concerns, ORA has worked with other FDA stakeholders and identified potential strategies to help address the concern without reducing inspection capacity. However, such strategies will not fully address the issues underlying these root causes, such as the amount of travel. For example, according to agency documents and officials, ORA is considering strategies to improve the travel planning process, which could reduce investigators’ travel stress and discomfort.[58] However, this would not affect the amount of travel required and its effect on work-life balance. Additionally, these potential strategies are still in development and thus lack action plans for implementation.
Leadership in ORA, CDER, and FDA more broadly is also collaborating to identify strategies to help meet inspection goals and improve overall inspection capacity, though these efforts do not address the root causes leading to investigator burnout and attrition. For example, according to an agency document, ORA and CDER are implementing a multi-year plan for CDER to conduct certain preapproval inspections instead of ORA, which could reduce the total burden of inspections on ORA. However, according to ORA officials, this would not result in reduced inspections and travel for individual investigators. Investigators would still be conducting the same number of inspections (and inspection-related travel) to meet other inspection goals, such as for surveillance inspections.
Given the need to balance current inspectional needs against the need to retain an experienced workforce, developing action plans to address attrition will likely require ORA, CDER, and other relevant stakeholders to continue to collaborate to identify strategies that appropriately balance these priorities. To successfully develop action plans that address the remaining root causes of attrition, FDA will need to identify the actions, resources, and any new authorities necessary to implement them. This will require an agency-wide effort with additional FDA stakeholders beyond ORA and CDER if implementing action plans requires changes to regulations or to current FDA or HHS policy or additional resources beyond what is currently available to ORA. For example, ORA officials stated that increasing the number of investigators could help meet inspection goals in the future, but ORA would need time and additional senior investigators in order to train them. According to officials, additional funding and resources to support training while ORA rebuilds its inspection workforce could help alleviate pressure on the current senior investigators, who are also needed to conduct inspections. However, officials told us that FDA does not have the resources to provide additional training resources.
While we recognize that addressing these travel, workload, and work-life balance issues could affect ORA’s ability to meet inspection goals, the continued loss of experienced investigators is already having this effect. Developing and implementing action plans can help FDA reduce investigator turnover and maintain an experienced investigator workforce. Continued collaboration between ORA, CDER, and other stakeholders in developing these action plans will help ensure that FDA identifies strategies that balance current inspectional needs against the need to retain an experienced workforce, as well as any actions, resources, or new authorities necessary to implement them. Lack of an experienced workforce increases the burden on senior investigators and further reduces FDA’s inspection resources, making it unable to fully meet its mission to oversee the global drug manufacturing supply chain.
FDA Used Certain Alternative Tools Extensively during the COVID-19 Pandemic and Plans Some Continued Use to Supplement In-Person Inspections
FDA used three types of alternative inspection tools
during the COVID-19 pandemic. During the pandemic, FDA expanded its use of
inspection reports from foreign regulators. The agency also used remote records
reviews extensively but used remote interactive evaluations relatively rarely.
Going forward, FDA plans to continue to use all three alternative tools, when
warranted, to supplement in-person inspections.
FDA Increased Use of Inspection Reports from Foreign Regulators during the COVID-19 Pandemic
|
Inspection Report Sharing
This means requesting and reviewing inspection reports from foreign regulators, including reports received via mutual recognition agreement. Source: GAO analysis of Food and Drug Administration information (text); Seventyfour/stock.adobe.com (photo). | GAO‑25‑106775 |

From fiscal years 2020 through 2023, FDA increased its use of inspection reports from European regulators with which FDA has a mutual recognition agreement in lieu of routine surveillance inspections of establishments manufacturing drugs already marketed in the U.S.[59] In fiscal year 2019, FDA substituted reports from inspections that European regulators conducted within their own country for about 100 FDA inspections. In the first year of the pandemic, fiscal year 2020, FDA substituted such inspection reports for over 160 FDA inspections. That total reduced in fiscal years 2021 and 2022 due to a decrease in inspection activity among all regulators, according to FDA officials.[60] However, when travel restrictions eased and European regulator domestic inspections rebounded in fiscal year 2023, FDA substituted almost 200 such reports for its own inspections.
FDA also used the added availability of reports of inspections conducted by certain European regulators in other countries during this period, although FDA decreased its use of them over time. Specifically, FDA substituted reports of inspections conducted by European regulators in other countries, such as India and China, for FDA inspections about 30 times in fiscal year 2021, but this decreased to less than five times in fiscal year 2023. FDA indicated this decrease was because European regulators had reduced their foreign inspection travel during that time and there were fewer inspection reports available.
FDA also used foreign regulator inspection reports to support the review of applications for new drugs or for marketed drugs with manufacturing changes. FDA used information from foreign regulators for review of drug applications over 120 times from fiscal year 2021 through fiscal year 2023.[61] During this time period, FDA reviewed European regulator inspection reports for this purpose over 100 times, while they reviewed inspection reports from other foreign regulators less than 20 times. FDA officials explained that using inspection information from regulators with whom FDA does not have a mutual recognition agreement is resource intensive, because each report is assessed on a case-by-case basis. For example, FDA once received an inspection report from a regulator that was three pages long with few details, which was insufficient for FDA’s needs.
Although FDA has increased its use of inspection reports from European regulators with which it has a mutual recognition agreement, the agency still may need to conduct its own inspections in their countries. For example, according to FDA officials FDA may determine an inspection report from a European regulator is not applicable if the scope of the inspection was different, or if FDA identifies risks that require an in-person inspection. FDA may also determine, on a case-by-case basis, that due to extenuating circumstances, certain inspections should be conducted by FDA, such as those initiated by information received from a confidential informant, or with a high likelihood of litigation. In addition, FDA officials explained that FDA regulates some products as drugs, such as sunscreens containing SPF and anti-cavity toothpaste, whereas they are regulated as cosmetics by European regulators. In the absence of an applicable report or other inspection results, FDA may request that the European regulator conduct an inspection on FDA’s behalf; however, this is dependent on the other regulator’s inspection capacity. FDA officials explained that a portion of these requests may remain unfulfilled by the time needed, in which case FDA must perform the inspection.
FDA Used Remote Records Reviews Extensively during the COVID-19 Pandemic and Changed Its Policies and Practices as the Pandemic Progressed
FDA used remote records reviews for two main purposes from fiscal year 2021 through 2023: 1) to gather information that would normally be obtained during an inspection and 2) to provide more targeted oversight of manufacturing establishments.
|
Remote Records Reviews
These include requesting and reviewing records and other information from the establishment under section 704(a)(4) of the Federal Food, Drug, and Cosmetic Act. Source: GAO analysis of Food and Drug Administration information (text); khunkornStudio/stock.adobe.com (photo). | GAO‑25‑106775 |

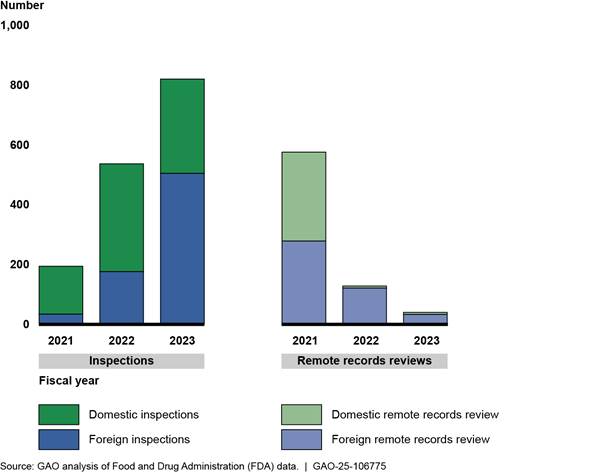
Gathering information normally collected during an inspection. While not equivalent to an inspection, FDA used these reviews to gather information it could use to inform decisions about drug approval or the scope of future surveillance inspections. Early in the COVID-19 pandemic, FDA used remote records reviews extensively in this way, with nearly 600 reviews in fiscal year 2021. (We previously reported that, prior to the COVID-19 pandemic, FDA used remote records reviews in a more limited capacity. Specifically, FDA used this tool to obtain information from 10 establishments that the agency would not routinely inspect because of travel warnings.[62]) However, this use of remote records reviews declined as FDA returned to routine domestic inspections in July 2021 and then later returned to routine foreign inspections in March 2022 everywhere except China. (See fig. 7.) The remote records reviews FDA conducted in fiscal year 2021 were nearly evenly split between foreign and domestic establishments. However, for fiscal years 2022 and 2023, more than two-thirds of FDA’s remote records reviews were related to foreign establishments—most frequently to those located in China and India.
Figure 7: FDA Inspections and Remote Records Reviews Used to Support Drug Application Review or Surveillance of Manufacturing Establishments, by Location, Fiscal Years 2021–2023
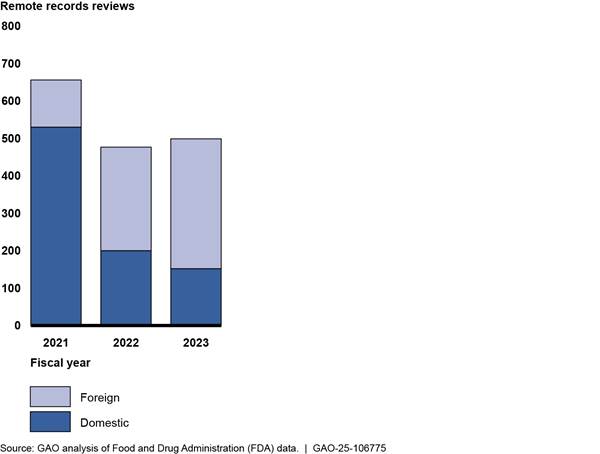
Providing more targeted oversight of manufacturing establishments. From fiscal years 2021 through 2023, FDA also used remote records reviews to request records and other information from establishments for more targeted reasons, as outlined below. The use of targeted remote records reviews remained relatively high across these three fiscal years, even after inspections resumed (see fig. 8).
Figure 8: FDA Remote Review of
Drug Manufacturing Establishment Records for a Targeted Reason, by Location,
Fiscal Years 2021–2023
According to FDA officials, the agency conducted targeted reviews for the following reasons.
· Triaging for potential inspections. According to officials, FDA used these reviews to determine whether an establishment was subject to an inspection before sending out investigators. Officials told us that FDA requested records from foreign and domestic establishments that had not previously received a human drug surveillance inspection.[63]
· Preparing for planned inspections. FDA officials told us that using these reviews in advance of a planned inspection can allow investigators to identify and focus on areas of concern during the subsequent inspection. FDA requested records from about 50 foreign and domestic establishments for this purpose.
· Investigating potential compliance issues. According to officials, FDA used these reviews to evaluate establishments’ compliance and take regulatory action as appropriate. This allowed the agency to help stop potentially contaminated drugs from entering the U.S. supply chain without conducting inspections. Based on these reviews, FDA issued warning letters and placed manufacturers on import alert based on their failure to respond to FDA’s request for records or based on inadequate responses.[64] (See table 5.)
Table 5: Examples of FDA’s Use of Remote Records Reviews to Investigate Potential Compliance Issues at Drug Manufacturing Establishments, Fiscal Years 2021–2023
|
Drug |
Potential compliance issue |
Use of remote records reviews |
|
Hand sanitizer |
In 2020, FDA identified a sharp increase in hand sanitizer products that were labeled to contain ethanol but tested positive for contamination with substances like methanol. Methanol can be toxic when absorbed through the skin and can be life-threatening when ingested. |
FDA issued about 700 records requests to hand sanitizer manufacturers to assess whether drug manufacturers were conducting required testing for contaminants and were in compliance with relevant FDA guidance. |
|
Oral liquid drugs, such as cough, allergy, and pain relief medications |
In 2022 and 2023, oral liquid drugs contaminated with diethylene glycol or ethylene glycol were associated with more than 300 deaths outside of the U.S., primarily among children under the age of 5. |
FDA issued about 170 records requests to assess whether drug manufacturers were conducting required testing to detect and prevent diethylene glycol contamination. |
|
Non-application sterile drugs, including eye drops and ointments |
In 2023, FDA identified contaminated over-the-counter eye drops that led to infections, partial loss of vision, and blindness. |
FDA issued two requests to drug manufacturers that had never been inspected to assess manufacturer controls to ensure product sterility and preservative formulations, according to FDA officials.a |
Source: GAO analysis of Food and Drug Administration (FDA) information. | GAO‑25‑106775
Note: Based on the failure of manufacturing establishments to respond to FDA’s requests for records or based on inadequate responses, FDA issued warning letters (which notify establishments that FDA may take enforcement action if violations are not promptly and adequately corrected) and placed manufacturers on import alert (which inform FDA staff that the agency has enough evidence to detain an establishment’s products that have been offered for entry into the U.S.)
aSome over-the-counter drug manufacturers can
legally market their products in the U.S. before FDA has conducted an
inspection to verify compliance with manufacturing requirements. In addition to
these two remote records reviews, FDA also conducted in-person inspections of
other manufacturers of these non-application sterile drugs, according to FDA
officials.
Over the course of the COVID-19 pandemic, FDA changed its remote records reviews’ policies and practices. These changes were in areas where industry stakeholders noted concerns about their experiences with remote records reviews, as outlined below.
· Defining the scope and rationale of requests. All six industry stakeholders we interviewed reported that it was challenging to understand the scope and purpose of FDA’s records requests. As a result, three industry stakeholders said establishments may provide a large volume of records in the hopes that what they provided would meet FDA’s needs. Three of the 10 FDA investigators we interviewed said reviewing the volume of records provided was a challenge, and three investigators told us that establishments provided the wrong documents in response to FDA’s requests. In January 2024, FDA updated its remote assessment draft guidance to clarify that, when making records requests, the agency would provide a sufficient description of the records requested, as well as a rationale for the request.[65]
· Providing document submission options. Two of the six industry stakeholders said submitting the requested records to FDA via email was challenging. Given the large number of documents provided, manufacturing establishments sometimes had to send multiple emails to FDA to avoid size limitations of email attachments. As the pandemic progressed, FDA expanded use of a file-sharing platform that made submitting records much easier, according to FDA officials.
·
Communicating about status and violations. All six of the
industry stakeholders we interviewed reported challenges understanding when
FDA’s review of the records was complete and whether the agency had identified
any violations. As a result, four stakeholders told us establishments were
uncertain about their regulatory compliance status. However, in mid-2021, FDA
changed this process to routinely provide a list of identified deficiencies at
the conclusion of the review, according to FDA officials. In addition, in
January 2024, FDA updated its remote assessment draft guidance to clarify that
if FDA does not have a closeout meeting at the conclusion of the remote
assessment, it will notify the establishment that the assessment is concluded
and share any pertinent information.[66]
FDA Rarely Used Remote Interactive Evaluations During the COVID-19 Pandemic
|
Remote Interactive Evaluations
These include the use of teleconference, livestream video, and screen sharing of data and documents with the establishment. Source: GAO analysis of Food and Drug Administration information (text); NIKCOA/stock.adobe.com (photo). | GAO‑25‑106775 |

FDA began using the new tool of remote interactive evaluations during the pandemic but used them rarely compared to other alternative tools. FDA conducted nine of these evaluations from fiscal years 2021 through 2023, all of them with foreign drug manufacturing establishments, according to FDA data. (See fig. 9.) All nine evaluations were either related to FDA’s review of a drug or biologics license application or related to FDA’s review of an emergency use authorization request.[67] In such instances, FDA used this tool to help assess manufacturing establishment risks identified in the agency’s review of the application or request.
Figure 9: FDA’s Use of Remote Interactive Evaluations of Human Drug Manufacturing Establishments, Fiscal Years 2021–2023
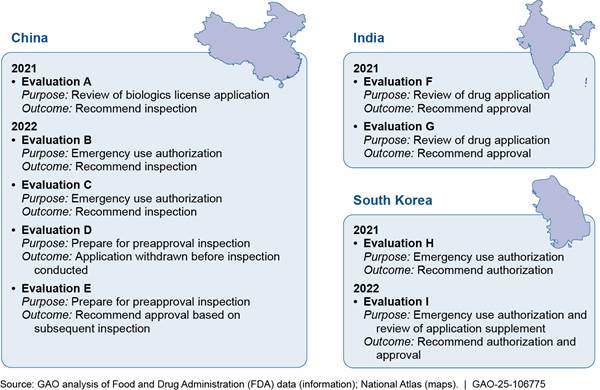
FDA officials described logistical and technical challenges that may hinder the usefulness of remote interactive evaluations.
· Logistical challenges. For evaluations conducted with establishments in Asian countries, FDA staff in the U.S. had to conduct them in the middle of the night to correspond with establishments’ normal working hours. Also, FDA needed interpreters to help conduct interviews for some remote interactive evaluations.
· Technical challenges. Audio and video quality, as well as Wi-Fi connectivity, were not always good. FDA officials explained that some areas within manufacturing establishments either didn’t have Wi-Fi or the Wi-Fi connection was impaired due to the nature of the facility, such as thickness of walls or interference from manufacturing equipment. In addition, they said that, because the establishment controls the video during a remote interactive evaluation, investigators can only view what the establishment shows them and are thus uncertain if they are getting a complete view of the establishment’s operations.
According to agency documents, FDA does not have the
authority to require either domestic or foreign establishments to participate
in a
remote interactive evaluation (unlike an in-person inspection), and this may
limit when they are used. The lack of authority to require participation may
limit any regulatory action FDA can take if an establishment refuses to
participate in a part of a remote interactive evaluation, according to FDA
officials. For example, if an establishment refused to allow an FDA investigator
to take a picture of a piece of equipment during an inspection conducted in
person, FDA could take regulatory action. However, FDA could not take such
action if an establishment refuses to shift the camera to focus on a particular
piece of equipment during a virtual tour. As of May 2024, FDA’s use of this
alternative tool in its oversight of drug manufacturing establishments has been
limited to assisting in the agency’s review of a drug application or emergency
use authorization, both situations in which the establishment has an interest
in participating in the evaluation.
This lack of authority to mandate that establishments participate in remote interactive evaluations may also reduce the efficiency of the remote records review alternative tool. All six industry stakeholders we interviewed agreed that the remote records review process would be more efficient if it included a real-time interactive component, such as a telephone call or virtual meeting. Most stakeholders said the remote records reviews their companies experienced were largely conducted via email, which two stakeholders said could result in multiple rounds of correspondence with FDA. In contrast, during an in-person inspection, FDA investigators ask establishments questions in real time as records are reviewed. According to all six industry stakeholders and four investigators, incorporating a real-time interactive component into a remote records review could ultimately increase the efficiency of this tool by, for example, ensuring that the establishment knows what records to provide and that investigators understand the records provided. However, according to FDA officials, incorporating such an interaction would turn the remote records review, which is mandatory, into a remote interactive evaluation, which is voluntary. As such, investigators were hesitant to do so as establishments could decline to participate, according to one investigator. FDA has requested authority to require remote interactive evaluations in each of its fiscal years 2023 through 2025 budget requests.
FDA Plans Continued Use of Alternative Tools When Warranted to Supplement Its Inspection Program
While in-person inspections will continue to be a primary oversight method, FDA has identified certain circumstances in which it will routinely use alternative tools—foreign regulator inspection reports, remote records reviews, and remote interactive evaluations. The agency also continues to consider other ways in which alternative tools may be used to supplement its inspection program.
We previously found that though FDA relied on alternative tools while inspections were paused due to the COVID-19 pandemic, the agency had not yet established future plans for these tools.[68] We recommended that FDA fully assess its alternative inspection tools and consider whether these tools or others could continue to provide the information needed to supplement regular inspection activities or help meet its drug oversight objectives when inspections are not possible. As noted above, FDA officials told us that FDA completed its assessment of the use of reports for inspections that European regulators conducted outside of Europe, expanding its use of this tool. In addition, in October 2021, FDA created a work group to review its remote assessment practices—that is, the use of remote records reviews and remote interactive evaluations—agencywide (including human and animal drugs, medical devices, tobacco, human foods, and biologics) and identify how such assessments could be used. Based on this review, the work group developed a July 2022 external guidance document for industry and an October 2022 internal procedural document for FDA staff. These documents outline the general circumstances in which, using a risk-based approach, FDA may consider using remote assessments and procedures for conducting them.[69] FDA officials told us that the agency generally has discretion to determine whether to conduct an inspection or use an alternative tool, based on the particular oversight need.
Based on this assessment, FDA officials described a number of specific instances in which the agency would use alternative tools more routinely to supplement the agency’s inspection program, as outlined below. Officials also described plans for potential expanded use of such tools.
· Inspection report sharing. According to FDA documents, the agency plans to continue to routinely use inspection reports from European regulators to support the review of drug applications and in lieu of surveillance inspections as appropriate, but the use of inspection information from other regulators may be limited, according to FDA officials. FDA plans to continue looking for opportunities to expand its use of reports from European regulator inspections conducted outside of Europe. However, FDA officials told us that the availability of such reports is limited by the volume of inspections these regulators conduct. For example, according to FDA officials, FDA conducts more inspections in China and India than other regulators. In addition, drug manufacturing establishments in those countries are more likely than in Europe to have distinct manufacturing lines or buildings that produce drugs for the European and U.S. markets separately. In such cases, FDA officials explained that inspections conducted by European regulators would not be suitable to FDA’s purposes.[70]
According to FDA officials, FDA has considered whether it should enter into mutual recognition agreements with additional regulators but has not yet begun the process with additional candidates due to current resources. To assess potential new regulatory partners, FDA considers its existing regulatory relationship with the regulator and the maturity of their regulatory framework, along with the cost and benefits of developing, assessing, implementing, and maintaining an agreement compared to the size of the industry, among other factors.[71] For example, China and India have nearly 40 percent of all foreign establishments manufacturing drugs for the U.S. market. However, according to FDA officials, the capacity of China and India to oversee drug manufacturing establishments in their own countries and the maturity of their regulatory frameworks are challenges to the establishment of mutual recognition agreements with these regulators. As a result, FDA has not yet pursued the establishment of such agreements with either regulator.[72]
· Remote records reviews. FDA officials told us that they do not plan for routine use of remote records reviews in lieu of surveillance inspections, unless travel is again limited. However, they described three specific situations in which they do plan to continue routinely using them: (1) to triage establishments to identify whether an inspection is warranted, (2) to investigate potential compliance issues, and (3) to support the agency’s review of drug applications. In all three cases, remote records reviews can help FDA make regulatory decisions without conducting an inspection. (Officials added that in all three instances, if an inspection was still needed after the use of the remote records review, FDA would conduct one.)
Officials also told us that FDA plans to consider whether to expand the use of remote records reviews in advance of certain inspections. FDA plans to request records in advance of some preannounced foreign surveillance inspections conducted by the dedicated foreign drug cadre in fiscal year 2025. Officials stated that this should help investigators focus on critical concerns once they are on site. As of May 2024, officials told us that FDA was in the process of drafting a proposal for this use, which will include metrics to evaluate its feasibility and determine whether to use it more broadly in the future.
· Remote interactive evaluations. FDA officials outlined two situations in which they are considering how to expand the use of remote interactive evaluations. First, officials told us that FDA plans to expand the use of this tool to gather new information to support a decision about a drug application when the listed establishment has a good compliance history.
FDA is also exploring the use of remote interactive evaluations in conjunction with a preapproval inspection to reduce the need for multiple inspections from different regulators. FDA is participating in a pilot program with other foreign regulators related to identifying best practices for collaborative hybrid inspections.[73] During a collaborative hybrid inspection, a regulator from one country performs an on-site inspection, while regulators from one or more other countries connect to the inspection remotely. According to the pilot implementation plan and related documents, the pilot is intended to identify best practices and technology for performing hybrid inspections. As of May 2024, FDA had participated in two pilot inspections, one as the on-site regulator and one as the remote participant. Based on the results of the pilot, FDA and other regulators will evaluate the feasibility of collaborative hybrid inspections more broadly, according to the pilot implementation plan.
These actions partially meet the intent of our recommendation for FDA to fully assess its alternative inspection tools and consider how they could be used in the future. To fully implement this recommendation, FDA should complete its planned assessment of its proposal to request records in advance of certain inspections and of the collaborative hybrid inspections pilot.
Remote assessments and workforce challenges. According to FDA officials, FDA is also considering the use of remote assessments—remote record reviews and remote interactive evaluations—to alleviate investigator shortages, though there may be limited benefits. While remote assessments reduce travel, according to FDA officials, such tools do not permit the full assessment of manufacturing practices that an inspection would allow. All 10 investigators and former investigators that we interviewed preferred in-person inspections to remote assessments except in limited circumstances, such as to assist in the agency’s review of a drug application when the establishment has a good compliance history. Further, remote assessments conducted to gather information that would normally be obtained during an inspection are largely conducted by the same investigators that conduct in-person inspections and take a similar amount of time, according to FDA officials. Given this resource overlap, officials told us that if FDA has a choice between spending the same amount of time on a remote assessment or conducting a surveillance inspection, the preferred choice is to spend the time onsite.
However, officials told us that FDA is considering the use of remote assessments in certain circumstances to help alleviate investigator shortages in two ways:
· Giving ORA investigators time to focus on certain types of inspections. FDA officials told us that in fiscal year 2024, CDER staff began leading remote assessments related to drug application reviews, rather than ORA investigators. This allowed FDA to collect certain information it needs to support decisions about drug applications, while freeing up ORA personnel time for surveillance and for-cause inspections.
· Reducing travel time and workload. While remote assessments take the same amount of time as surveillance inspections (according to FDA officials), the latter also include travel time. Officials said that performing remote assessments in lieu of certain inspections or as a supplement to other inspections could help alleviate concerns from
investigators about the amount
of travel and the workload required for inspections. According to FDA
officials, use of remote assessments, either temporarily or more routinely,
could thus help with retention.
However, FDA officials told us they were still considering how to use such
assessments to help address investigator retention.
Selected Foreign Regulators Vary in Their Reliance on Alternative Tools to Oversee Drug Manufacturing
All seven of the selected foreign regulators we interviewed varied in the extent to which they rely on alternative tools to substitute or supplement in-person inspections in other countries.[74] The capacity to conduct inspections and the use of alternative tools are affected by the level of resources available to each regulator, according to a report by the National Academies of Sciences, Engineering, and Medicine.[75] Member states of the European Union, like FDA, are among the most highly resourced regulators, and European Union officials reported that member states rely more heavily on conducting foreign inspections, while using alternative tools as a supplement. Officials from Japan also reported relying more on conducting foreign inspections compared to alternative tools. In contrast, the other regulators that we interviewed reported relying less on conducting foreign inspections and using these tools as a primary oversight mechanism. During the COVID-19 pandemic, all seven regulators increased their reliance on alternative tools, including using inspection results from other regulators and conducting remote record reviews, and six of them introduced remote interactive evaluations.
|
GMP Certificates Food and Drug Administration (FDA) officials explained
that there are nuanced differences in regulations between countries that
could mean an establishment with a Good Manufacturing Practice (GMP)
certificate from another regulator may not meet U.S. requirements. For example,
the U.S. requires under 21 C.F.R. 211.42, that penicillin products be
manufactured in separate facilities to prevent cross-contamination and treats
other beta lactams similarly. However, according to FDA officials, some other
regulators do not have this same requirement. Source: GAO analysis of FDA information. | GAO‑25‑106775 |
Sharing inspection results. All seven of the selected foreign regulators use inspection results from other regulators to oversee drug manufacturing in other countries, but five of the regulators use them in a majority of oversight decisions while the other two use them less frequently. Regulators use inspection reports and Good Manufacturing Practices (GMP) certificates of approval from other regulators via inspection sharing agreements, including mutual recognition agreements and other arrangements, to make oversight decisions.[76] For example, a regulator may assess a shared inspection report to determine if it meets the regulator’s oversight needs. In addition, all of the regulators explained that they can rely on GMP certificates of approval from trusted foreign regulators in certain circumstances. For example, a regulator may rely on a GMP certificate without having to conduct an in-person inspection or may postpone their inspection to a later date. GMP certificates are issued by regulators to establishments at the conclusion of a successful inspection. The establishment can then share them with other regulators assessing compliance. FDA does not issue GMP certificates as a part of its regulatory framework or use them in lieu of conducting an inspection. (See sidebar.) As described earlier, FDA instead uses inspection reports from foreign regulators along with other information about the facilities to determine whether a facility is in compliance with manufacturing requirements.
Officials from the European Union reported that member states use inspection sharing agreements whenever possible, but that they still rely on in-person foreign inspections as a major oversight tool. In contrast, five of the regulators that we interviewed relied more heavily on shared inspection results, including from mutual recognition agreement partners or other trusted foreign regulators. For example, Australia, Canada, New Zealand, Switzerland, and the United Kingdom reported using such information in a majority of their oversight decisions for foreign manufacturers. These regulators cited factors such as a limited number of inspection staff and the efficient use of limited resources as reasons for the reliance on shared inspection results. The seventh regulator, Japan, does not make oversight decisions based solely on information obtained from shared inspection results and instead uses this information in combination with an inspection, conducted either in-person or through a review of establishment records.
All of the regulators we interviewed noted the benefits of shared inspection results including for reducing duplication of inspections, directing inspection resources to higher-risk areas, and reducing the burden on industry. While shared inspection results are a critical source of information for all of the regulators, their availability relies on the capacity of other regulators to conduct the relevant inspection. For example, regulators noted that fewer inspection results were available during the COVID-19 pandemic, when all regulators reduced or paused foreign inspection travel.
Remote review of establishment records. All seven of the foreign regulators we interviewed reported using remote review of records and other information from drug manufacturing establishments to inform oversight decisions but varied in the extent to which they use the information in lieu of an inspection or a shared inspection report.[77] All of the regulators reported that they regularly use remote review of establishment records combined with inspection information shared by other regulators to assess manufacturers. Officials from the European Union and the United Kingdom reported they did not otherwise rely on document review in lieu of an inspection or shared inspection report. These officials explained that remote review of establishment records was only a component of remote interactive evaluations. Finally, two of the seven regulators may use remote review of establishment records in lieu of certain types of low-risk inspections (e.g., certain types of manufacturing changes, for manufacturers with a positive history of inspections).
Four of the foreign regulators we interviewed said that a primary benefit of remote review of establishment records is the flexibility it can afford when travel is a challenge, including restrictions due to circumstances such as security risks and pandemics or due to resource limitations. However, three of those regulators noted that despite these benefits, there are challenges to using this tool including that it can be time consuming. In addition, two regulators told us that during the COVID-19 pandemic they expanded their use of remote review of establishment records, for example requesting new types of information that they would normally review during an inspection.
Remote interactive evaluations. All but one of the selected foreign regulators used remote interactive evaluations to oversee foreign drug manufacturing in specific circumstances, such as when travel is not possible or a critical drug supply may be affected. None of the regulators we interviewed routinely used remote interactive evaluations—which include virtual meetings, real-time document review using screensharing, and video tours of the facility—prior to the COVID-19 pandemic. All of the regulators except for New Zealand used remote interactive evaluations at some point during the pandemic. All of the regulators that used remote interactive evaluations during the pandemic told us that they reduced their use of it when inspection travel resumed.
Five of the foreign regulators we interviewed described similar challenges using remote interactive evaluations, comparable to those expressed by FDA. This included adjusting to time zone differences, challenges viewing certain manufacturing areas or details via video, and technological limitations with Wi-Fi signal. Two regulators also cited the inability to observe staff body language, such as gestures and physical signs, that may indicate stress or nervousness. In addition, two of the regulators expressed interest in exploring the feasibility of using a hybrid approach in which one regulator uses a combination of remote and inspection activities. For example, the regulator may first obtain documentation from the establishment that would normally be reviewed during an inspection, then conduct interviews via video teleconference, and finally schedule a brief and targeted inspection to assess processes that they were unable to verify remotely.
Conclusions
FDA’s inspections of drug manufacturing establishments are a key tool to ensure the safety and quality of drugs marketed in the U.S. The agency has adapted to growing globalization through increased foreign inspections of the drug supply chain. However, this has presented challenges for its investigator workforce and attrition has left it with a less experienced workforce and less capacity to conduct needed inspections. As a result, the agency continues to conduct many fewer inspections than it did at its peak in fiscal year 2016. If investigator attrition is not addressed, the continued turnover and hiring of new investigators may increase the burden on senior investigators, as such investigators help train new staff. This would further reduce the inspection resources available for FDA, making it unable to fully meet its mission to oversee the global drug manufacturing supply chain.
FDA is aware of these challenges and stakeholders from ORA, CDER, and agency leadership are engaged in efforts to identify strategies to address them. Its recent efforts have identified the root causes of investigator attrition, and the agency has taken some steps in response. For example, FDA has taken steps to increase pay and has begun to consider how alternative tools could help address these concerns, either temporarily as FDA rebuilds its investigator workforce or as a permanent supplement to its inspection program. These steps may provide additional incentives to investigators and provide FDA with options to maintain oversight despite its reduced capacity. However, the agency has not identified or documented strategies that can address the remaining root causes of travel, workload, and work-life balance concerns. These strategies will need to go beyond identifying opportunities to increase overall inspection capacity and also account for the burnout experienced by individual investigators. We have identified FDA workforce challenges over several reports. The persistence of this problem is integral to our designation of FDA’s oversight of the increasingly global medical product supply chain as a high-risk area. Given these challenges, developing and implementing action plans to address these remaining root causes and ensuring collaboration among ORA, CDER, and other relevant stakeholders, will help the agency identify opportunities to balance current inspectional needs while retaining an experienced workforce.
Recommendation for Executive Action
The Commissioner of FDA should ensure that FDA develops and implements action plans that address attrition caused by issues with investigator travel, workload, and work-life balance. In doing so, ORA, CDER, and other relevant stakeholders should collaborate to identify strategies that balance current inspectional needs against the need to retain an experienced workforce and identify any necessary actions, resources, or new authorities. (Recommendation 1)
Agency Comments
We provided a draft of this report to HHS for review and comment. We also provided excerpts of this report to the seven foreign regulators that we interviewed for their review and comment. HHS and four of the seven foreign regulators provided technical comments, which we incorporated as appropriate.
We also received written comments from HHS, which are reproduced in appendix I. In its comments, the agency concurred with our recommendation that it develop and implement action plans that address investigator attrition. HHS stated that FDA plans to establish a committee under the office of the FDA Commissioner that will be responsible for developing an action plan to comprehensively address investigator attrition issues and will include representation from all affected departments. HHS further noted that this committee will collaborate with appropriate stakeholders to integrate initiatives that are already underway into a holistic strategy to address attrition.
We are sending copies of this report to the appropriate congressional committees, the Secretary of Health and Human Services, the Commissioner of the Food and Drug Administration, and other interested parties. In addition, the report will be available at no charge on the GAO website at https://www.gao.gov.
If you or your staff have any questions about this report, please contact me at (202) 512-7114 or DeniganMacauleyM@gao.gov. Contact points for our Offices of Congressional Relations and Public Affairs may be found on the last page of this report. GAO staff who made key contributions to this report are listed in appendix II.
Mary Denigan-Macauley
Director, Health Care


GAO Contact
Mary Denigan-Macauley, Director, Health Care, (202) 512-7114 or deniganmacauleym@gao.gov.
Staff Acknowledgements
In addition to the contact named above, William Hadley (Assistant Director), Katherine L. Amoroso (Analyst-in-Charge), Sonia Chakrabarty, Taneeka Hansen, Ariel Jona, David Jones, Noelle Miesfeld, Laurie Pachter, and Dan Ries made key contributions to this report.
FDA Inspections of Drug Manufacturing
Drug Safety: FDA Should Take Additional Steps to Improve Its Foreign Inspection Program. GAO‑22‑103611. Washington, D.C.: January 7, 2022.
COVID-19: Critical Vaccine Distribution, Supply Chain, Program Integrity, and Other Challenges Require Focused Federal Attention. GAO‑21‑265. Washington, D.C.: January 28, 2021.
Drug Safety: Preliminary Findings Indicate Persistent Challenges with FDA Foreign Inspections. GAO‑20‑262T. Washington, D.C.: December 10, 2019.
Drug Safety: FDA Has Improved Its Foreign Drug Inspection Program, but Needs to Assess the Effectiveness and Staffing of Its Foreign Offices. GAO‑17‑143. Washington, D.C., December 16, 2016.
Drug Safety: FDA Has Conducted More Foreign Inspections and Begun to Improve its Information on Foreign Establishments, but More Progress is Needed. GAO‑10‑961. Washington, D.C.: September 30, 2010.
Food and Drug Administration: Overseas Offices Have Taken Steps to Help Ensure Import Safety, but More Long-term Planning Is Needed. GAO‑10‑960. Washington, D.C.: September 30, 2010.
Drug Safety: Better Data Management and More Inspections Are Needed to Strengthen FDA’s Foreign Drug Inspection Program. GAO‑08‑970. Washington, D.C.: September 22, 2008.
Drug Safety: Preliminary Findings Suggest Recent FDA Initiatives Have Potential, but Do Not Fully Address Weaknesses in Its Foreign Drug Inspection Program. GAO‑08‑701T. Washington, D.C.: April 22, 2008.
Drug Safety: Preliminary Findings Suggest Weaknesses in FDA’s Program for Inspecting Foreign Drug Manufacturers. GAO‑08‑224T. Washington, D.C.: November 1, 2007.
Food and Drug Administration: Improvements Needed in the Foreign Drug Inspection Program. GAO/HEHS‑98‑21. Washington, D.C.: March 17, 1998.
Other FDA Inspections Related to Medical Products
Clinical Research: FDA Should Evaluate Its Efforts to Recruit and Retain Its Inspection Workforce. GAO‑24‑106383. Washington, D.C.: February 22, 2024.
Institutional Review Boards: Actions Needed to Improve Federal Oversight and Examine Effectiveness. GAO‑23‑104721. Washington, D.C.: January 17, 2023.
COVID-19: Additional Actions Needed to Improve Accountability and Program Effectiveness of Federal Response. GAO‑22‑105051. Washington, D.C.: October 27, 2021.
GAO’s Mission
The Government Accountability Office, the audit, evaluation, and investigative arm of Congress, exists to support Congress in meeting its constitutional responsibilities and to help improve the performance and accountability of the federal government for the American people. GAO examines the use of public funds; evaluates federal programs and policies; and provides analyses, recommendations, and other assistance to help Congress make informed oversight, policy, and funding decisions. GAO’s commitment to good government is reflected in its core values of accountability, integrity, and reliability.
Obtaining Copies of GAO Reports and Testimony
The fastest and easiest way to obtain copies of GAO documents at no cost is through our website. Each weekday afternoon, GAO posts on its website newly released reports, testimony, and correspondence. You can also subscribe to GAO’s email updates to receive notification of newly posted products.
Order by Phone
The price of each GAO publication reflects GAO’s actual cost of production and distribution and depends on the number of pages in the publication and whether the publication is printed in color or black and white. Pricing and ordering information is posted on GAO’s website, https://www.gao.gov/ordering.htm.
Place orders by calling (202) 512-6000, toll free (866) 801-7077,
or
TDD (202) 512-2537.
Orders may be paid for using American Express, Discover Card, MasterCard, Visa, check, or money order. Call for additional information.
Connect with GAO
Connect with GAO on Facebook, Flickr, X, and YouTube.
Subscribe to our RSS Feeds or Email Updates. Listen to our Podcasts.
Visit GAO on the web at https://www.gao.gov.
To Report Fraud, Waste, and Abuse in Federal Programs
Contact FraudNet:
Website: https://www.gao.gov/about/what-gao-does/fraudnet
Automated answering system: (800) 424-5454 or (202) 512-7700
Congressional Relations
A. Nicole Clowers, Managing Director, ClowersA@gao.gov, (202) 512-4400, U.S. Government Accountability Office, 441 G Street NW, Room 7125, Washington, DC 20548
Public Affairs
Sarah Kaczmarek, Managing Director, KaczmarekS@gao.gov, (202) 512-4800, U.S.
Government Accountability Office, 441 G Street NW, Room 7149
Washington, DC 20548
Strategic Planning and External Liaison
Stephen J. Sanford, Managing
Director, spel@gao.gov, (202) 512-4707
U.S. Government Accountability Office, 441 G Street NW, Room 7814, Washington,
DC 20548
[1]Drugs are defined to include, among other things, articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease and include components of those articles. See 21 U.S.C. § 321(g)(1)(B), (D). An active pharmaceutical ingredient includes, among other things, any substance that is intended to provide pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease. See 21 C.F.R. § 207.1. In this report, we refer both to drug products—drugs in their finished dosage forms—and to active pharmaceutical ingredients as “drugs.”
[2]U.S. Food & Drug Administration, Center for Drug Evaluation and Research, Fiscal Year 2023 Report on the State of Pharmaceutical Quality (June 2024).
[3]See GAO, Drug Safety: FDA Should Take Additional Steps to Improve Its Foreign Inspection Program, GAO‑22‑103611 (Washington, D.C., Jan. 7, 2022).
[4]The agency resumed normal operations for domestic inspections in July 2021 and began resuming normal operations for foreign inspections in March 2022, according to FDA officials.
[5]See, for example, GAO‑22‑103611 and GAO, High-Risk Series: Efforts Made to Achieve Progress Need to be Maintained and Expanded to Fully Address All Areas, GAO‑23‑106203 (Washington, D.C.: Apr. 20, 2023). For a list of related reports, see the Related GAO Products page at the end of this report.
[6]See GAO, Drug Safety: Preliminary Findings Indicate Persistent Challenges with FDA Foreign Inspections, GAO‑20‑262T (Washington, D.C.: Dec. 10, 2019).
[8]Pub. L. No. 117-328, § 3614, 136 Stat. 4459, 5872 (2022).
[9]The inspection data we used for some prior reports came from FDA’s Field Accomplishments and Compliance Tracking System. Since January 2015, data on human drug manufacturing inspections was entered into a new data system called eNSpect. The data used in this report comes from eNSpect.
[10]See GAO, State Department: Additional Actions Needed to Address IT Workforce Challenges, GAO‑22‑105932 (Washington, D.C.: July 12, 2022). This report identified selected leading practices for recruitment and retention. From among the retention leading practices, we focused on leading practices related to employee morale, as our initial audit work on the drug investigator workforce identified morale as the most relevant factor given FDA’s workforce issues and existing efforts.
[11]Specifically, we reviewed documents from and interviewed the Association for Accessible Medicines, Bulk Pharmaceuticals Task Force, Consumer Healthcare Products Association, Pharmaceutical Research and Manufacturers of America, Biotechnology Innovation Organization, and Pharma & Biopharma Outsourcing Association, which represent manufacturers of generic drugs, active ingredients, over-the-counter drugs, brand-name drugs, and contract manufacturers, respectively.
[12]The names of the regulators are Therapeutic Goods Administration (Australia), Health Canada (Canada), Pharmaceuticals and Medical Devices Agency (Japan), MedSafe (New Zealand), Swissmedic (Switzerland), Medicines and Healthcare products Regulatory Agency (United Kingdom), and European Medicines Agency (European Union). (The European Medicines Agency coordinates drug oversight activities for drugs authorized centrally among the 27 member states within the European Union and between the European Union and other countries. For the purposes of this report, we refer to the European Medicines Agency as a regulator.)
[13]Two of the regulators provided written responses, rather than being interviewed.
[14]Our analysis focused on inspections related to the drug approval process or inspections conducted to determine an establishment’s ongoing compliance with laws and regulations in the manufacture of human drugs already marketed in the U.S. FDA conducts additional drug inspections that are beyond the scope of our review, such as to determine whether drug manufacturers are submitting to FDA, as required, complete and accurate data on adverse drug experiences associated with marketed drugs; inspections conducted for the President’s Emergency Plan for AIDS Relief; and inspections of clinical trial sites, compounding pharmacies, and medical gas manufacturers. FDA also conducts inspections of other products such as biologics regulated by FDA’s Center for Biologics Evaluation and Research, veterinary medicines, and medical devices, which are outside the scope of our review.
[15]FDA’s sampling and testing program assesses product quality, rather than manufacturing quality, and does not confirm adherence to quality manufacturing standards. Thus, while sampling and testing is an oversight tool, such activities were excluded from this study.
[16]Current GMPs provide for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. See 21 U.S.C. § 351(a)(2)(B); 21 C.F.R. parts 210, 211, 212. FDA considers nearly all drug establishment inspections to include an assessment of current good manufacturing practices.
[17]At times, FDA may conduct an inspection that combines both preapproval and surveillance inspection components in a single visit to an establishment. Most combined inspections occur when FDA conducts a surveillance inspection at an establishment where a preapproval inspection is also being conducted.
[18]See 21 U.S.C. § 360(h)(3), (4).
[19]See GAO‑22‑103611.
[20]The six strategies developed by the work group include: 1) utilize alternative hiring authorities and compensation to recruit new and retain current investigators specializing in foreign drug inspections, 2) conduct ongoing and specialized recruitment efforts, 3) standardize training, including on the job training on foreign inspections, 4) maximize retention incentives, such as cash bonuses, 5) travel process improvements, and 6) enhance opportunities for development and diversity of work for investigators interested in specializing in foreign inspections.
[21]In order to enter into such an agreement, FDA must determine that the foreign regulatory authority has the capability to conduct inspections that meet FDA requirements. Food and Drug Administration Safety and Innovation Act, Pub. L. No. 112-144, § 712, 126 Stat. 993, 1072 (2012) (codified at 21 U.S.C. § 384e).
[22]For the purposes of this report, when we refer to European regulators, we are referring to the 27 European Union member regulators that are part of the mutual recognition agreement between the European Union and the U.S., plus the United Kingdom and Switzerland, with which the U.S. has separate mutual recognition agreements. The mutual recognition agreement between the U.S. and the European Union went into force in 2017, followed by agreements with the United Kingdom in 2021 and Switzerland in 2023. FDA completed capability assessments of all of the European Union member’s regulatory authorities individually by July 2019, after which FDA could use inspection reports from the regulators in lieu of an FDA inspection.
[23]See GAO, COVID-19: Critical Vaccine Distribution, Supply Chain, Program Integrity, and Other Challenges Require Focused Federal Attention, GAO‑21‑265 (Washington, D.C.: Jan. 28, 2021).
[24]The mutual recognition agreement between FDA and European regulators allows FDA to recognize inspections conducted by European regulators outside of Europe. However, before the COVID-19 pandemic this authority had not been implemented because FDA had not yet completed the requisite capability assessment. The interim policy expansion in March 2020 enabled FDA to recognize these inspections before the capability assessments were officially completed. As of July 2024, FDA had formally completed assessments of European regulators to recognize inspections conducted outside of Europe, with the exception of Switzerland, according to FDA. (FDA’s assessment of the Swiss regulator’s inspections conducted outside of Switzerland is pending.)
[25]The Pharmaceutical Inspection Co-operation Scheme is a consortium of 56 regulators that facilitates cooperation and networking between regulators, develops common standards for drug manufacturing inspection practices, and aims to harmonize inspection procedures through training opportunities for inspectors. Of the 56 members of the consortium, FDA has existing sharing agreements with 43 regulators through mutual recognition agreements and confidentiality commitments, according to FDA.
[26]Pub. L. No. 112-144, § 706, 126 Stat. at 1068 (codified at 21 U.S.C. § 374(a)(4)).
[27]In fiscal year 2019, for example, this included establishments in Colombia, Egypt, Israel, Mexico, Pakistan, and Saudi Arabia, among others.
[28]See U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, Center for Veterinary Medicine, Remote Interactive Evaluations of Drug Manufacturing and Bioresearch Monitoring Facilities During the COVID-19 Public Health Emergency: Guidance for Industry (April 2021). FDA first published this guidance in April 2021 pursuant to the COVID-19 public health emergency. In October 2023, FDA withdrew the April 2021 version of this guidance, and issued an updated draft guidance.
[29]FDA announced in March 2020 that, in light of the COVID-19 pandemic, the agency would temporarily not conduct any foreign or domestic inspections other than those deemed mission-critical. FDA identifies mission-critical inspections on a case-by-case basis by considering many factors related to the public health benefit of patients having access to the product subject to inspection, as well as considering the safety of its inspection staff and employees of the establishment to be inspected. See GAO‑22‑103611. In July 2020, FDA resumed other higher priority domestic inspections in locations of lower COVID-19 risk. FDA reported that it resumed routine, non-mission critical domestic inspections in July 2021 and non-mission critical foreign inspections by March 2022.
[30]Full fiscal year 2024 inspection data were not available at the time of our review.
[31]In 2022, we reported that, according to FDA officials, staff located in the agency’s China and India offices conducted certain prioritized inspections during fiscal year 2021. While FDA reported that it resumed routine foreign inspections in March 2022, officials stated that some inspections in China were delayed because of COVID-19 restrictions.
[32]Preapproval inspections are conducted before approving a drug to be marketed in the U.S., while for-cause inspections are conducted to investigate specific issues. Routine surveillance inspections are conducted periodically at establishments manufacturing drugs already marketed in the U.S. to assess compliance. FDA prioritized inspections during the COVID-19 pandemic based on factors such as the effect of the product’s availability on public health and the public health risk or benefit posed by the potential inspection site. For example, for-cause inspections were considered mission critical or prioritized when there was evidence of serious adverse events or to follow-up on a product recall because product safety or quality could be at risk. Preapproval inspections were prioritized to support the approval decision of products critical to FDA’s public health mission.
[33]Some fiscal year 2023 inspections have not yet been classified by FDA, and therefore the overall classification rates for fiscal year 2023 could change once those inspections receive a final classification.
[34]In our January 2022 report, we found that FDA planned on implementing two pilot programs to address these challenges. However, we reported that those efforts were delayed by the COVID-19 pandemic and that the agency had not finalized the pilots’ designs. We recommended that FDA incorporate the following leading practices into the design of both of its pilot programs: 1) establish well defined, appropriate, clear, and measurable objectives; 2) articulate an assessment methodology that details the type and source of the information necessary to evaluate the pilot and the methods for collecting that information; 3) develop an evaluation strategy that defines how the information collected will be analyzed to evaluate the pilot’s implementation and performance; 4) assess the scalability of the pilot design to inform whether and how to implement a new approach in a broader context; and 5) ensure appropriate stakeholder communication at all stages of the pilot. FDA agreed with our recommendations. See GAO‑22‑103611.
[35]Unannounced inspections include those announced on short notice—those in which establishments receive notice of the inspection 72 hours or less before the inspection begins. Establishments may receive much less than 72 hours’ notice, including less than 1 hour.
See Food and Drug Omnibus Reform Act of 2022, Pub. L. No. 117-328, § 3615, 136 Stat. 5807, 5873.
[36]We previously reported that some investigators prefer conducting team inspections as it helps reduce risks to their personal safety. In our current work, two of 10 investigators and former investigators we interviewed also expressed a preference for team inspections as it made the intense workload associated with foreign inspections more reasonable. However, FDA officials told us that sending multiple investigators on all inspections would reduce the number of inspections FDA is able to conduct.
[37]If FDA identifies serious deficiencies in relation to a foreign establishment, the agency may place the drug products or establishment on an import alert, which informs FDA staff and the public that the agency has enough evidence to detain an establishment’s products that have been offered for entry into the U.S.
[38]The total number of vacancies for November 2021 includes only the vacancies for the general pool and the foreign offices and does not include foreign cadre vacancies. According to FDA data and officials, while there were 10 open positions in the cadre in November 2021 these positions were not counted as additional vacancies because they were already counted in the total authorized positions for the general pool. As of June 2024, FDA now separates the cadre authorized positions from the general pool authorized positions.
[39]See GAO‑22‑103611.
[40]In our prior report, GAO‑22‑103611, we counted certain investigator positions as filled for which a new hire had a scheduled start date but had not yet begun work. The data FDA provided for this report, including revised data for November 2021, include only those investigators officially onboard as of that date. Additionally, since our prior report, FDA has determined that certain positions which only occasionally conduct inspections should be excluded from the count of onboard investigators that FDA provided for the previous report.
[41]As a strategy to improve recruitment into the foreign cadre, FDA offers current investigators in the general pool a 6-month detail to the cadre. Of the five investigators who accepted a detail in 2022, three have since become permanent cadre members; officials noted that prior to creating the 6-month detail they were not receiving as many applications for cadre positions. As noted above, in our prior report we counted certain investigator positions as filled for which an investigator had a scheduled start date but had not yet begun work. The data FDA provided for this report, including revised data for November 2021, include only those investigators officially onboard with the cadre as of that date.
[42]Investigators in the foreign offices are recruited from qualified investigators in the general investigator pool or the foreign cadre for 2-year assignments but can chose to extend their assignment up to 6 years per office location. During the time that ORA investigators are posted full-time overseas, they are detailed to the Office of Global Policy and Strategy, which manages the foreign offices, among other duties. In addition to 2-year assignments, FDA staff can also be assigned to the foreign offices on temporary duty assignments for up to 120 days. For example, during fiscal year 2023, two investigators served temporary details in China, and one served a detail in India.
[43]One additional investigator had accepted a tentative offer for a position in the China office but was not yet in China as of June 2024. For the India office, two additional investigators accepted tentative offers but were not yet in the country as of June 2024. For a prior report, officials told us that obtaining the necessary security and medical clearances and other prerequisites for serving in a foreign office can take 9 to 12 months.
[44]This includes two investigators who took assignments to a foreign office and thus were detailed to the Office of Global Policy and Strategy and not ORA. It also includes 11 investigators who took other, non-investigator positions within the Office of Pharmaceutical Quality Operations, the ORA office that conducts drug manufacturing inspections, which could include promotions.
[45]All new investigators are initially hired by ORA and begin their careers in the general pool. Qualified investigators can then be recruited from the general pool by ORA into its foreign cadre or by the Office of Global Policy and Strategy for assignments to the foreign offices. When investigators leave the foreign cadre or positions in the foreign office at the end of their assignment or for other reasons, they may return to the general pool of investigators. Therefore, investigators leaving these foreign focused positions do not necessarily increase overall investigator attrition.
[46]GAO‑22‑103611. Our recommendation focused on recruiting and retaining investigators who specialize in foreign inspections, particularly the foreign drug cadre and the investigators in FDA’s foreign offices. In response, FDA established a work group that identified six strategies. While some of the strategies are specific to recruiting foreign specialists, others are also relevant to the issue of attrition in the general pool.
[47]The Federal Employee Viewpoint Survey measures employees’ perceptions of whether, and to what extent, conditions characterizing successful organizations are present in their agencies. The survey has been conducted annually by the Office of Personnel Management since 2010. It measures topics such as job and pay satisfaction, inclusivity in the workplace, and agency conditions that lead to employee engagement, which is an indicator of employee morale.
[48]We have also previously reported that investigators identified challenges, particularly with travel and workload. See for example GAO‑22‑103611 and GAO‑20‑262T.
[49]Officials additionally described challenges with travel planning, such as restrictions stemming from federal regulations or policies related to air travel and challenges with receiving official passports in time for the planned trip, which can increase travel stress and discomfort and potentially result in a trip being rescheduled.
[50]Participants in FDA’s focus group also had concerns related to safety while traveling, including access to medical services and health insurance coverage while in a foreign country. According to agency documents, FDA does not have authority to provide medical assistance and evacuation insurance for investigators on foreign travel, and, as a result, an investigator would be responsible for the cost of any medical emergencies while on foreign travel. FDA requested this authority as part of its fiscal year 2025 budget request.
[51]Salary data was only available for a portion of investigators who transferred to other FDA centers. Further, FDA did not have salary data for investigators who left FDA for a different federal agency or for a non-government position, according to officials.
[52]We previously reported that, according to FDA officials, members of the dedicated foreign drug cadre can receive up to 15 hours of overtime per week during an overseas trip to complete inspection-related work. Five investigators we interviewed for a previous report stated they worked long hours to complete their work.
[53]Hiring authority under Title 21 was originally provided by the 21st Century Cures Act, a law enacted in 2016 which, among other things, provided special pay authorities to HHS to facilitate FDA’s recruitment and retention of medical product staff. Pub. L. No. 114-255, § 3072(a), 130 Stat. 1033, 1134 (2016) (codified at 21 U.S.C. § 379d-3a). The Food and Drug Omnibus Reform Act of 2022 expanded this authority to apply to more positions and offices within FDA. Pub. L. No. 117-328, § 3624, 136 Stat. 5807, 5879.
[54]ORA’s Title 21 action plan prioritizes recruiting new investigators and, on a case-by-case basis, converting select mission-critical staff at risk of leaving. The remaining drug investigators will have their positions converted in later fiscal years. Investigators will not receive a universal pay increase when their position is converted, as FDA did not receive additional funding to support such an increase, but the conversion will provide staff with a higher maximum salary and thus the potential for salary growth.
[55]While FDA has action plans to improve training, agency documents show that FDA anticipates new hire training will continue to be a challenge due to limited resources, specifically the limited number of senior investigators available to train new investigators and planned cuts to the training budget.
[56]See GAO‑22‑105932.
[57]U.S. Office of Personnel Management, Workforce Planning Guide, ES-03483-11/2022 (Washington, D.C.: November 2022).
[58]ORA is leading a working group, started in October 2023, that is examining potential solutions to challenges with the travel planning process, such as issues with obtaining official passports and airline seat selections. Several of the proposed strategies will require policy changes outside of FDA’s control, including by HHS and the Department of State, which issues official passports. The working group’s efforts are still ongoing.
ORA has also requested changes to the deadlines for investigators to complete inspection reports, according to officials. Such changes could improve investigators’ workloads after inspections. At the time of our review the agency had not identified specific plans or timelines for implementation of these changes.
[59]For the purposes of this report, when we refer to European regulators, we are referring to the 27 European Union member regulators that are part of the mutual recognition agreement with the U.S., plus the United Kingdom and Switzerland, which have separate mutual recognition agreements with the U.S. Mutual recognition agreements between FDA and European regulators allow FDA to recognize inspections conducted by European regulators deemed capable of conducting inspections that meet U.S. requirements.
[60]FDA officials explained that, at the beginning of the COVID-19 pandemic, there were a limited number of inspections that had been conducted by European regulators after FDA had determined their inspection reports were eligible for substitution but before the pandemic had limited inspection activities by all regulators. As such, there were a limited number of inspection reports that FDA could request. The number of eligible inspection reports increased as foreign regulator inspection activity resumed.
[61]In addition to European regulators, FDA used inspection reports from regulators that are among the 56 members of the Pharmaceutical Inspection Cooperation Scheme, including those with which FDA has a Confidentiality Commitment (such as Australia, Canada, Japan, and South Africa).
[62]See GAO‑21‑265.
[63]FDA data shows that from fiscal year 2021 through 2023, more than 200 of the more than 500 records requests made to triage establishments for potential inspection resulted in FDA determining that the establishment did not actually need to be inspected because, for example, it was not manufacturing drugs for the U.S. market. Thus, FDA removed these establishment from its catalog of establishments subject to inspection.
[64]FDA may issue warning letters to establishments manufacturing drugs for the U.S. market that are in violation of applicable U.S. laws and regulations and thus may be subject to enforcement action if violations are not promptly and adequately corrected. In addition, if FDA identifies serious deficiencies in relation to a foreign establishment, the agency may place the drug products or establishment on an import alert, which informs FDA staff that the agency has enough evidence to detain an establishment’s products that have been offered for entry into the U.S.
[65]Federal law requires FDA to provide a sufficient description of and a rationale for the request. 21 U.S.C. § 374(a)(4)(A). See also U.S. Department of Health and Human Services, Food and Drug Administration, Office of Regulatory Affairs, Office of Food Policy and Response, Office of Combination Products, Center for Biologics Evaluation and Research, Center for Drug Evaluation and Research, Center for Devices and Radiological Health, Center for Food Safety and Applied Nutrition, Center for Tobacco Products, Center for Veterinary Medicine, Conducting Remote Regulatory Assessments: Questions and Answers – Draft Guidance for Industry (January 2024).
[66]FDA officials explained that not all types of remote records reviews would result in a closeout meeting and sharing of deficiencies. First, reviews conducted to triage establishments for potential inspection would not result in a list of deficiencies if FDA identified that the establishment does not produce drugs for the U.S. market. Second, FDA would not share deficiencies identified in records reviewed in advance of an inspection as such deficiencies would be further examined during the subsequent inspection.
[67]Biologics are generally derived from living material, such as the human body or a microorganism, and are generally more complex than chemically synthesized drugs. FDA generally licenses biologics for marketing in the U.S. through approval of a biologics license application. An emergency use authorization allows for the temporary use of medical products without FDA approval or licensure, provided certain statutory criteria are met. See 21 U.S.C. § 360bbb-3. For more information on emergency use authorizations, see GAO, COVID-19: Federal Efforts Accelerate Vaccine and Therapeutic Development, but More Transparency Needed on Emergency Use Authorizations, GAO‑21‑207 (Washington, D.C.: Nov. 17, 2020).
[68]See GAO‑21‑265.
[69]According to FDA guidance, factors that may be considered when making the risk-based determination of whether to use a remote assessment tool include establishment location, inspection history, complexity of product and process, and travel restrictions. FDA issued an updated version of its external guidance in January 2024. See U.S. Department of Health and Human Services, Conducting Remote Regulatory Assessments.
[70]European regulator inspections are funded directly by the inspected establishment, and that funding may not cover the cost of any additional inspection activities that are specific to FDA, such as an inspection of a separate manufacturing building.
[71]U.S. Food and Drug Administration, Framework for Mutual Recognition Agreements and Arrangements Relating to Drugs, Staff Manual Guide 9121 (Effective February 13, 2023).
[72]FDA also considered regulators with more mature regulatory frameworks, such as Norway and Iceland, however, according to FDA officials, those countries do not have enough establishments manufacturing drugs for the U.S. market to warrant pursuing such agreements.
[73]According to FDA officials, FDA is co-lead (along with the European Medicines Agency) of the working group implementing this pilot as part of its participation in the International Coalition of Medicines Regulatory Authorities.
[74]GAO interviewed six regulators and the European Medicines Agency. The regulators were: Australia (Therapeutic Goods Administration), Canada (Health Canada), Japan (Pharmaceuticals and Medical Devices Agency), New Zealand (MedSafe), Switzerland (Swissmedic), and the United Kingdom (Medicines and Healthcare products Regulatory Agency). The European Medicines Agency coordinates drug oversight activities for drugs authorized centrally among the 27 member states within the European Union and between the European Union and other countries. For the purposes of this report, we refer to the European Medicines Agency as a regulator.
[75]National Academies of Sciences, Engineering, and Medicine, Regulating Medicines in a Globalized World: The Need for Increased Reliance Among Regulators (Washington, DC: The National Academies Press, 2020).
[76]GMP certificates are issued by a country’s regulatory authority to a manufacturer to provide confirmation of that manufacturer’s compliance with GMP requirements. Such certificates are generally valid for 3 years, but that can be adjusted under special circumstances.
[77]While new drug applications include a review of documentation submitted to a regulator by the manufacturer or sponsor, here we are referring to remote document reviews that are performed either in lieu of or as a component of an inspection or for the purposes of gathering information normally collected during an inspection to inform decisions about drug approval or the scope of future inspections.